










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La fibrosis quística (FQ) ha mejorado la supervivencia y la calidad de vida en las últimas décadas, pasó de ser considerada una enfermedad fatal en edades tempranas de la vida a ser una enfermedad crónica. Esto es debido a la puesta en marcha de unidades de fibrosis quística especializadas y a la utilización de nuevas modalidades terapéuticas, que se han traducido en un diagnóstico precoz, una mejoría del estado nutricional y disminución de las infecciones respiratorias. Según estudios realizados la frecuencia en Cuba es de 1 × 9 000 nacidos vivos.1
Hace 80 años Anderson describió la "fibrosis quística del páncreas”, enfermedad generalmente fatal en el primer año de la vida. En Cuba desde la década de los años 50 del siglo xx comenzaron los primeros reportes de esta enfermedad. El primer caso registrado en Cuba fue diagnosticado por necropsia en 1953. En la década de los setenta un equipo médico orientado por el Profesor Manuel Rojo Concepción profundiza en el estudio, diagnóstico y tratamiento de la FQ. En 1974 se crea la Comisión Nacional de Fibrosis Quística (CCFQ), con varias metas, la primordial de ellas, la mejor atención al paciente y sus familiares.1) Durante más de cuarenta años esta Comisión ha trabajado y obtenido importantes resultados.
En 1963 se crea en el Hospital Pediátrico “William Soler” el Servicio de Enfermedades Respiratorias para el seguimiento de afecciones respiratorias agudas y crónicas. Desde el año 1974 pertenece a La Comisión Nacional de Fibrosis Quística donde se discuten por un grupo de expertos y aprueban o no los casos sospechosos, con el propósito de detectar tempranamente la enfermedad y aplicar la terapéutica adecuada para alcanzar mayor estándar de supervivencia y calidad de vida en los niños que sufren FQ.
En el 2007 existían en Cuba 230 pacientes con esta enfermedad. En el 2017 la cifra era de 279 pacientes, el 13 % mayor de 30 años, con un promedio de vida de 18,1 años.2)
La FQ es una enfermedad multisistemica, que se hereda con carácter autosómico recesivo, condiciona un daño pulmonar progresivo desde la etapa de lactante.3 Es causada por la alteración de un único gen localizado en el brazo largo del cromosoma 7, gen regulador de la conductancia transmembrana de la fibrosis quística, que codifica una proteína alterada, la proteína CFTR, siglas en inglés. La enfermedad aparece cuando los dos genes FQ del paciente presentan mutaciones que hacen que la proteína no exista o que tenga alterada su función.4
La proteína que codifica el gen CFTR se comporta como un canal de cloro regulado por AMPc y las mutaciones de este gen dan lugar a un defecto en el transporte del cloro en las células epiteliales del aparato respiratorio, hepatobiliar, gastrointestinal, reproductor, páncreas y de las glándulas sudoríparas. Las glándulas mucosas afectadas elaboran secreciones excesivamente viscosas, por lo que a veces se denomina la enfermedad como mucoviscidosis.4
Se establece el diagnóstico de FQ en presencia de al menos una característica fenotípica de la enfermedad (enfermedad sinopulmonar crónica, alteraciones nutricionales, síndromes de pérdida de sal, ausencia bilateral de conductos deferentes asociado o no a insuficiencia pancreática), junto con una concentración de cloro en el sudor ≥ 60 mmol/L, según el método que se use. La evolución clínica de esta enfermedad es variable.5
La mitad de las personas con FQ son homocigotos para ∆F508. La mayor parte de las demás personas tiene una copia de la mutación ∆F508 y otra alteración del gen de la FQ. Se describen otras mutaciones frecuentes como. G542X, R1162X, R334W, R553X, N1303K, entre otras. Han sido identificadas más de 1932 mutaciones.6)
El patrón y la frecuencia con que se presentan estas mutaciones dependen del origen étnico del paciente y se han encontrado frecuencias variables en las distintas poblaciones.7
Aunque al nacer los pacientes con FQ tienen los pulmones normales, la afectación progresiva de las vías respiratorias y su resultante: el cor pulmonale, es la causa de cronicidad y muerte en más del 90 % de los casos.8 Sin embargo, por la multiplicidad de órganos y sistemas a los que afecta, la FQ es una enfermedad muy compleja que requiere ser abordada de forma integral en su tratamiento y seguimiento.9)
En etapas tempranas de la vida puede aparecer colonización por Staphilococcus aureus y Haemophilus influenzae, seguido de Pseudomonas aeruginosa, que es el germen que más frecuentemente se aísla en los enfermos con FQ. La prevalencia difiere según los distintos países y está relacionada con la edad, aparece en 30 % de los pacientes entre los 2 y 5 años de edad, pero a partir de los 18 años están colonizados aproximadamente el 80 %. Además, últimamente hay indicios de que hasta 30 % de los pacientes menores de 2 años pueden tener cultivos positivos a Pseudomonas aeruginosa, que generalmente se adquiere a partir de microorganismos presentes en el medio ambiente, también se ha descrito la transmisión de cepas de paciente a paciente o su adquisición durante los ingresos hospitalarios.10,11
El propósito de los autores de este trabajo es describir las características de los pacientes con fibrosis quística y el seguimiento de los vivos durante 40 años.
MÉTODOS
Se realizó un estudio descriptivo longitudinal retrospectivo sobre FQ en niños, y para ello, se revisaron todas las historias clínicas de los pacientes diagnosticados por tener sospecha clínica de esta enfermedad en el Hospital Pediátrico Universitario “William Soler” a partir de enero 1977 hasta diciembre de 2017. Se incluyeron en el estudio todos los niños con diagnóstico positivo aprobados por la Comisión Nacional de Fibrosis Quística desde el nacimiento hasta 18 años 11 meses y 29 días, con 96 pacientes que conformaron el universo y la muestra.
Las variables estudiadas fueron: sexo, color de la piel, edad del diagnóstico y edad actual, el estado nutricional según los percentiles de peso para la talla de acuerdo con las tablas cubanas de crecimiento y desarrollo, expresados como: delgado (entre 3 y < 10 percentil), eutrófico (entre 10 y < 90 percentil), sobrepeso (entre 90 y 97 percentil) y obeso (> 97 percentil); forma clínica que sugirió la sospecha diagnóstica, aislamiento de gérmenes en secreciones obtenidas por hisopado faríngeo profundo, mutación identificada, complicaciones y fallecidos en el período de estudio.
Se obtuvieron los datos de las historias clínicas y con ellos se confeccionó una base de datos automatizada en software Excel y se expresaron los datos en porcentajes. El Comité de Ética del Hospital Pediátrico aprobó la investigación.
RESULTADOS
Durante cuatro décadas se han diagnosticado en el Hospital Pediátrico Universitario “William Soler”, 96 niños con FQ, 30 se siguen en otras instituciones, 9 han pasado a seguimiento en hospital de adultos, 30 fallecieron y actualmente se siguen en la unidad de fibrosis quística del citado hospital 27 pacientes.
El 60,4 % de ellos se diagnosticó antes del año de edad, el diagnóstico fue disminuyendo con la edad: después de los 10 años solo se diagnosticó 4,2 % de los pacientes. El 62,5 % de los niños fibroquisticos son del sexo masculino, y 88,5 %, de color de piel blanco.
En la tabla 1 se aprecia que la forma de presentación de la enfermedad que predominó fue la respiratoria pura (38 pacientes 39,6 %), seguida de la mixta respiratoria/digestiva (37 pacientes 38,5 %).
Tabla 1- Pacientes diagnosticados en la Unidad de Fibrosis Quística del Hospital Pediátrico Universitario “William Soler” (n= 96)
| Variable | n | % |
|---|---|---|
| Sexo | ||
| Masculino | 60 | 62,5 |
| Femenino | 36 | 37,5 |
| Apariencia racial | ||
| Blanco | 85 | 88,5 |
| Mestizo (mezcla racial) | 11 | 11,5 |
| Negro | - | - |
| Edad del diagnóstico | ||
| Menor de 1 año | 58 | 60,4 |
| 1-4 años | 23 | 25,0 |
| 5-9 años | 10 | 10,4 |
| Más de 10 años | 4 | 4,2 |
| Formas clínicas al diagnóstico | ||
| Digestiva | 19 | 19,8 |
| Mixta (respiratoria/digestiva) | 37 | 38,5 |
| Respiratoria | 38 | 39,6 |
| Perdedora de sal | 2 | 2,1 |
| Total | 96 | 100,0 |
Actualmente se siguen en la Unidad de Fibrosis Quística 27 pacientes, que al igual que el total de los niños diagnosticados en el período analizado, la mayoría 77,8 % se diagnosticó antes del año. Cuando se hizo el análisis de la edad actual vimos que el 40,8 % son adolescentes mayores de 15 años (tabla 2).
De los pacientes que se siguen en consulta, cuando se hizo el diagnóstico de FQ, 55,6 % eran desnutridos y el 25,9 % delgados; en el análisis actual observamos que no hay desnutridos y el 74,1 % de los niños son eutróficos (tabla 2).
Tabla 2- Pacientes con seguimiento actual (n= 27). Edad del diagnóstico / edad actual y valoración nutricional al diagnóstico/actual

Pc: percentil.
La mutación genética ∆F508 predominó (70,3 %): Homocigótico (48,1 %), el resto de los pacientes fueron portadores de otras mutaciones como ∆F508/Heterocigótico (con mutación no conocida hasta el momento) 3 pacientes (11,1 %), ∆F 508/G85E en 2 pacientes (7,4 %), el resto ∆F 508 /G542X, G542X/Heterocigótico, G542X/R553X, R553X/R334W, R553X/Heterocigótico con 1 paciente respectivamente; 4 pacientes a pesar de tener clínica y electrólitos en sudor positivos no tuvieron mutaciones de las estudiadas en Cuba (tabla 3).
La relación entre las manifestaciones clínicas que sugirieron el diagnóstico de la enfermedad y la mutación genética, (genotipo/fenotipo), refleja que los pacientes con mutación ∆F508/Homocigótico tuvieron manifestaciones clínicas tanto digestivas como respiratorias y que 2 pacientes tuvieron al nacimiento íleo meconial (tabla 3).
Tabla 3- Pacientes con seguimiento actual (n= 27). Relación entre la mutación genética y las manifestaciones clínicas
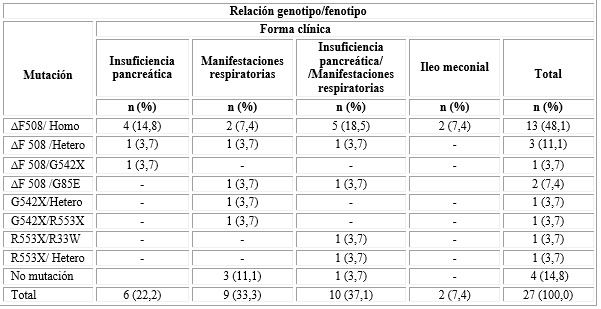
Homo: Homocigótico; Hetero: Heterocigótico.
No hubo complicaciones en el 40,7 % de los pacientes estudiados, la mayoría de las complicaciones fueron respiratorias: 37,0 % bronquiectasias, 29,6 % pansinusitis, 1 paciente presentó aspergilosis broncopulmonar y hemoptisis, 3 pacientes (11,1 %) poliposis nasal con necesidad de tratamiento quirúrgico; el 14,8 % de los niños tuvo complicaciones hepáticas, deformidad torácica y acropaquias respectivamente (tabla 4).
Tabla 4- Pacientes con seguimiento actual (n=27). Complicaciones
| Complicaciones* | n | % |
|---|---|---|
| Sin complicaciones | 11 | 40,7 |
| Complicaciones respiratorias | ||
| Bronquiectasias | 10 | 37,0 |
| Aspergiliosis broncopulmonar | 1 | 3,7 |
| Poliposis nasal | 3 | 11,1 |
| Hiperreactividad bronquial | 6 | 22,2 |
| Pansinusitis | 8 | 29,6 |
| Otras complicaciones | ||
| íleo meconial operado y vivo | 2 | 7,4 |
| Complicaciones hepáticas | 4 | 14,8 |
| Deformidad torácica | 4 | 14,8 |
| Acropaquias | 4 | 14,8 |
| Escroto agudo con tto. quirúrgico | 2 | 7,4 |
| Apendicitis complicada | 1 | 3,7 |
| Trastorno de atención e hiperactividad | 3 | 11,1 |
*Un paciente puede tener más de una complicación.
La mayoría de los aislamientos microbiológicos son antes de los 4 años de edad. En el primer aislamiento se detectó Pseudomonas aeruginosa en las secreciones respiratorias bajas en 8 niños menores de 1 año (29,6 %) de los enfermos, entre 1-4 años se aisló Staphilococcus aureus en 5 niños (18,5 %), Pseudomonas aeruginosa y Pseudomonas mucoide 4 pacientes (14,8 %), respectivamente y Pseudomonas aeruginosa/ Staphilococcus aureus 2 pacientes (7,4 %). No se aislaron bacterias en 5 niños (18,5 %). La mayoría de los fallecimientos ocurrieron antes del año 2000 (25 niños) con predominio de los menores de 1 año, las causas fueron: 6,2 % por íleo meconial y el resto, bronconeumonías, cirrosis hepática, obstrucción bronquial y cor pulmonar crónico. Después del 2000 solo murieron 5 niños por cor pulmonar crónico y bronconeumonía y de ellos 4 alcanzaron la adolescencia. En este período se salvaron los 2 pacientes diagnosticados con íleo meconial al nacimiento (Fig.).
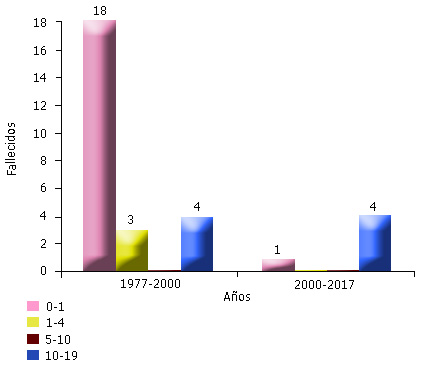
DISCUSIÓN
En esta investigación se encontró predominio del sexo masculino y el color de piel blanco. Se corresponde con la casuística cubana actual donde el 60,5 % de los pacientes FQ son del sexo masculino.2
Bautista y otros,12) en un estudio de 26 años describe que el 60 % de 48 niños se diagnosticaron antes del año de edad, 57,8 % del sexo masculino y 84.4 % de color de piel blanco, datos que coinciden con la casuística de este trabajo. Entre las formas clínicas que sugieren el diagnóstico predominan las formas mixtas digestivo-nutricional y respiratoria, al igual que en este trabajo.
En el 2011 en Asturias se publica en 45 pacientes fibroquisticos de todas las edades, diagnóstico antes del año de edad en 55,5 %, las características clínicas que sugieren la sospecha diagnóstica son las manifestaciones digestivas o nutricionales, seguido de las manifestaciones respiratorias en 15 % de los pacientes, sin embargo ellos encontraron a diferencia de este estudio, un predominio en el sexo femenino 57,8 %. 13)
Fuentes y otros,14 caracterizan a los niños fibroquisticos durante el primer año de vida y registran que el 79,5 % presentan manifestaciones pulmonares típicas, predominio en varones y desnutridos 62,5 % de sus casos. Para esta esta edad notifican aislamiento de Pseudomonas aeruginosa en 68,8 % de los niños, datos cercanos a la investigación presente.
Encontramos un incremento del diagnóstico de la enfermedad en los menores de un año, resultados superiores a los publicados, en el 2009 en Cuba, por Razón y otros,1 en 234 niños fibroquísticos, el diagnóstico fue antes del año de edad en el 49,5 %; y en el 13,4 % el diagnóstico se realizó a partir de los 16 años. En Santiago de Cuba, en el oriente del país, Guzmán y otros15 en el 2008, publican que 41,2 % de sus diagnósticos se hacen antes de los 5 años de edad.
En un estudio colombiano en 25 pacientes, 15 se diagnosticaron entre los primeros meses y los cinco años de edad; sin embargo, un gran porcentaje es diagnosticado en una edad tardía, aspecto que favorece el rápido avance y daño sistémico de la enfermedad.16
El árbol bronquial del paciente con FQ constituye un nicho ecológico favorable para el sobrecrecimiento de los microorganismos. El proceso de inflamación inicial, mediado en parte como consecuencia de las mutaciones en el gen CFTR, tiende a exacerbarse por la presencia de los microorganismos que terminan por colonizar de forma crónica la superficie mucosa y ejercen un claro deterioro de la función pulmonar.17
Los microorganismos que colonizan la vía aérea de los pacientes con FQ presentan una secuencia temporal relativamente establecida y asociada a la edad del paciente.17)
Durante las primeras etapas de la vida, las infecciones víricas propias de la infancia (también en el individuo no afecto de FQ) pueden provocar la denudación del epitelio pulmonar y favorecer la colonización bacteriana recurrente y el estado local de inflamación crónica. Se ha demostrado que algunos virus (Adenovirus y Coronavirus) y también determinadas bacterias (Mycoplasma pneumoniae y Chlamydophila pneumoniae) estimulan el sistema fagocítico, favorecen la descamación del epitelio y la atracción de los neutrófilos. Con ello se beneficia la respuesta inflamatoria presente en el tracto respiratorio, que puede evidenciarse incluso antes de que se aíslen los patógenos clásicos y característicos de la FQ.17)
Tras este período inicial, la colonización más frecuente es la causada por Staphylococcus aureus (Sa) y Haemophilus influenzae. Streptococcus pneumoniae coloniza también la mucosa respiratoria en las primeras etapas, pero su presencia no es más frecuente que en los niños de igual edad sin FQ; no obstante, presentan unos perfiles de resistencia mayores que en estos. Sa es a menudo el patógeno que inicia el proceso de colonización crónica que caracteriza a los pacientes con FQ.18 Recientemente, han aumentado las descripciones de aislamiento de Sa resistentes a la meticilina (SAMR). Conforme avanza la edad del paciente y la progresión de la enfermedad, decrece la colonización por Sa y aumenta el aislamiento de Pseudomona aeruginosa (Pa), que se incrementa de forma gradual hasta convertirse en el patógeno más frecuente en la edad adulta.17 En esta investigación se encontró Pa, en primer aislamiento en edades tempranas de la vida seguido de Sa.
La Pa se ha asociado al deterioro progresivo e irreversible de la función pulmonar, la persistencia de recuentos elevados y el cambio del morfotipo no-mucoide a mucoide se relacionan con más exacerbaciones y peor función pulmonar. La infección endobronquial crónica por este microorganismo es la causa más importante de morbilidad y mortalidad en estos pacientes.19,20Haemophilus influenzae aparece con menor frecuencia, junto con otras especies, como: Burkholderia cepacia, Achromobacter xylosoxydans o Stenotrophomonas maltophilia.21,22
Fuentes y otros,23 en un estudio sobre fallecidos por fibrosis quística encuentran que el 27 % estaba colonizado por Pa antes del año de vida, cifra cercana a la de esta investigación.
La primera mutación descrita, y la más frecuente a nivel mundial, es la ∆F508, pero existen otras mutaciones específicas cuya frecuencia varía entre los distintos grupos étnicos. En España se ha descrito una frecuencia media de la ∆F508 de entre 50-60 % del total de los cromosomas estudiados y la segunda en frecuencia es la G542X, con un 4-8 %, seguida por la A1303Lys con 2-4 %.17) En este estudio informamos predominio de la mutación ∆F508 seguido de la mutación G542X, no encontramos en esta población la A1303Lys, Bautista12 y Guzmán15 en Cuba tampoco han publicado esta última mutación. Las mutaciones que encontramos se corresponden con las informadas en Cuba en el 2008.1) Guzmán15 en el 2011 en Santiago de Cuba, informa predominio de la mutación ∆F508/Homocigótico (68,75 %), así como también de la presentación respiratoria (23,5 %), seguida muy de cerca por la mixta (20,6 %), datos estos coincidentes con los de la actual investigación.
La mutación G542X es común en los países del Mediterráneo, la frecuencia más alta se ha encontrado en Islas Baleares con 16,7 %, la frecuencia en la población mundial es de 4,4 % (Francia 3,1 %, Italia 4,8 %, España 7,7 %, Cuba 6%). La mutación N1303K, está presente alrededor del mediterráneo y alcanzó su mayor frecuencia (17,2 %) en Tunisia.24Bonilla16 en encuesta a cuidadores en Colombia, encuentra poca confirmación del diagnóstico con prueba genética, además los cuidadores consideran que el diagnóstico de la enfermedad fue tardío.
La importante morbilidad y mortalidad de esta enfermedad está relacionada con la afectación pulmonar y sus complicaciones, que son responsables del 95 % de los fallecimientos de los pacientes. En la década de los años 50, los pacientes fallecían antes de los dos años, en la década de los años 70, la mediana de supervivencia se incrementó hasta los 15 años. En el 2013, la Cystic Fibrosis Foundation (CFF) publica en su registro anual una mediana de supervivencia de 40,7 años. El aumento tan importante de la supervivencia de estos pacientes, en los últimos años, es debido a una serie de factores, entre los que ha contribuido de forma determinante la implantación de los programas de cribado neonatal y la instauración de una terapia precoz y agresiva.17
Las complicaciones que se presentaron en nuestros pacientes son, en su mayoría, por el deterioro pulmonar. El compromiso pulmonar progresivo deriva en hipoxemia e hipercapnia con cor pulmonar en etapa terminal, estos datos coinciden con lo publicado por Fuentes.23 Dentro de las complicaciones respiratorias menos frecuentes encontramos la hemoptisis, la poliposis nasal, el hipocratismo digital y la aspergilosis broncopulmonar, que corroboran lo publicado en la literatura revisada.17
Los enfermos con FQ desarrollan cambios inflamatorios de la mucosa rinosinusal que puede llevar a engrosamiento, acumulación de líquido y poliposis y se convierte en un reservorio de gérmenes patógenos de difícil llegada para los antibióticos. Se sabe que, aún en portadores de una mutación de fibrosis quística, la prevalencia de rinosinusitis es más alta que en aquella población no portadora.25,26 En esta investigación se encontró 11 % de los niños con pólipos nasales que necesitaron tratamiento quirúrgico y el 29,6 % con pansinusitis.
Ningún paciente del estudio presentó neumotórax y se describe que aparece en el 5-8 % de los pacientes con FQ, especialmente en aquellos con afección pulmonar moderada-grave.5
En el estudio realizado en Costa Rica, en el 2009, las complicaciones identificadas son: el prolapso rectal, la colelitiasis, la hepatopatía, la cirrosis hepática, la hemoptisis, la diabetes mellitus y el hipocratismo digital.27
El compromiso severo del hígado es, probablemente, la segunda causa de mortalidad en esta enfermedad. Se plantea que afecta entre 17-25 % de los pacientes, puede presentarse desde un hígado graso hasta cirrosis y condicionar hipertensión portal, várices esofágicas y sangrado digestivo de difícil manejo. La lesión hepática más grave es una cirrosis biliar focal, que puede evolucionar a una cirrosis biliar multilobular.28 Actualmente se siguen cuatro pacientes con afectación hepática. Entre los fallecidos en las 4 décadas, 2 casos presentaron cirrosis hepática, otros autores publican menor casuística.23
Castilla,9 en Perú informa que la causa de fallecimiento es, en la mayoría de los casos, por insuficiencia respiratoria asociada a sepsis (72 %). En nuestro seguimiento en 40 años, fallecieron 14,5 % de los niños con diagnóstico de bronconeumonía, 6,2 % por cor pulmonare crónico e íleo meconial, respectivamente. Los últimos diagnósticos de íleo meconial en el recién nacido sobrevivieron.
En el Taller Nacional de Fibrosis Quística, Cuba 2017, se expuso que las tasas medias anuales ajustadas de mortalidad según año desde el 1987 descendieron desde aproximadamente 1,5 por 100 000 habitantes a 0,7 en los últimos ocho años.2
Al concluir el trabajo, se comprobó que en 40 años de seguimiento la mayoría de los pacientes se diagnosticaron antes del primer año de vida. Es frecuente el aislamiento de Pseudomona aeruginosa en etapa de lactante, casi la mitad de los pacientes no tienen complicaciones, se logró mejorar el estado nutricional y disminuir la mortalidad.
Se considera importante continuar estos estudios, para realizar un diagnóstico precoz y seguimiento adecuado de estos pacientes.

