










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La Enfermedad de Niemann-Pick Tipo C (NP-C) es una rara enfermedad neurovisceral, producida por un severo trastorno lisosomal por acumulación de lípidos de herencia autosómica recesiva, causada entre el 95-98 % de los casos por la mutación del gen NP-C1 y el resto por la del gen NP-C2.1 El gen NP-C1 determina la proteína transportadora del colesterol intralisosomal NPC1 y su mutación más frecuente es la NP-C 111061T. La acumulación del colesterol dilata y disfunciona los lisosomas de las células y conlleva a variadas presentaciones clínicas dentro de un rango de síntomas viscerales, neurológicos y psiquiátricos.2 La expresión fenotípica de las variantes patogénicas de los genes del NP-C1 y del NP-C2 tiene un amplio rango, desde el comienzo fetal temprano con hydrops fetales hasta la demencia progresiva en adultos,2,3,4,5,6,7 y afecta un caso de cada 100 000 a 120 000 nacidos vivos.8).
En la última década varios estudios de cohortes han aportado conocimientos sobre la sintomatología y la historia natural de esta enfermedad.8 La enfermedad de NP-C puede presentarse a cualquier edad, aquellos que comienzan sus síntomas entre 2 meses y 2 años tienen síntomas principalmente viscerales que incluye esplenomegalia, hepatomegalia inexplicable, íctero neonatal prolongado e hiperbilirrubinemia directa, los cuales preceden al inicio de los signos neurológicos. La edad de comienzo de las primeras manifestaciones neurológicas y su progresión usualmente define la severidad de la enfermedad. Las manifestaciones neurológicas y psiquiátricas varían con la edad de comienzo de los síntomas.9 En la lactancia los síntomas incluyen:
Retardo de la adquisición de las habilidades motoras.
Retardo del lenguaje.
Regresión de habilidades del neurodesarrollo previamente adquiridas.
Hipotonía central.
Ataxia.
Cataplejía gelástica.
En el inicio juvenil incluyen:
Problemas escolares.
Deterioro intelectual.
Parálisis supranuclear de la mirada conjugada vertical.
Dismetría.
Ataxia cerebelosa.
Disartria o disfagia progresivas.
Crisis epilépticas.
En los adolescentes y adultos aparecen:
Lo heterogéneo y con frecuencia no específico de las manifestaciones clínicas del NP-C explica el frecuente retraso durante años de su diagnóstico.10
Recientemente se ha elaborado un instrumento clínico, el Índice de Sospecha (IS) del NP-C, que basado en síntomas y signos del paciente, tiene una alta sensibilidad en la sospecha clínica de esta enfermedad.11,12,13
Diagnóstico
El diagnóstico del NP-C dado la rareza y heterogeneidad clínica puede retrasarse durante años. En los últimos años se han ido perfeccionando los métodos clínicos, histológicos, de estudios por microscopía electrónica, bioquímicos y de genética molecular para su diagnóstico.8) Ejemplo de ellos son la tinción de filipin en cultivos de fibroblastos así como marcadores lisosomales del tipo LAMP1 (proteína 1 de membrana asociada al lisosoma) que participa en el transporte del colesterol desde los lisosomas a otros compartimentos celulares1 y los estudios genéticos que ) pueden precisar el tipo de mutación presente.2A pesar de ser esta una enfermedad neurodegenerativa fatal, en los últimos años han surgido un grupo de estrategias, con el objetivo de modificar el curso de la misma, como es el caso del miglustac que hace más lenta su progresión aunque tiene efectos secundarios que en ocasiones obliga a suspender su uso.1,14,15,16,17,18,19
El objetivo de este trabajo es contribuir al conocimiento de esta rara enfermedad neurovisceral progresiva de curso fatal.
PRESENTACIÓN DEL CASO
Paciente femenina, blanca, de 7 años de edad, procedente de la provincia de Guantánamo, hija de padres jóvenes, no consanguíneos, madre hipertensa, padre sano. La paciente ingresa por primera vez a la edad de dos años por retardo en la adquisición de habilidades del neurodesarrollo, fundamentalmente motoras y de lenguaje, así como crisis epilépticas. Evolutivamente presenta dificultades progresivas para la marcha, con trastornos del equilibrio, pérdida de la habilidad para agacharse hasta perder la marcha, con regresión del lenguaje adquirido, así como crisis epilépticas de varios tipos. Durante un ingreso por cuadro febril se detecta esplenomegalia, que fue a mayor evolutivamente. Posteriormente comienza a presentar caídas bruscas al piso cuando reía (cataplejía gelástica) y dificultad para tragar, sobre todo los sólidos.
Examen físico a la edad de 7 años y otros exámenes
Se tomaron los siguientes datos de la paciente a los 7años e edad: peso: 20 kg. Talla: 120 cm. Peso/edad: 90 percentil. Talla/edad: >97 percentil. Peso/talla: entre el 10 y 25 percentil. Macrocefalia. Circunferencia de la cintura (CC) 53 cm, mayor del 97 percentil. Facies inexpresiva. Boca entreabierta. Mala oclusión dentaria. Abdomen globuloso. Se palpa esplenomegalia de aproximadamente 4 cm, superficie lisa, no dolorosa. Hígado rebasa 1 cm el reborde costal. Paciente consciente, pero no se comunica con el examinador. Disminución del trofismo y del tono muscular. Reflejos osteotendinosos presentes y simétricos. Parálisis de la mirada vertical.
Electroencefalograma
Actividad epileptiforme focal intercrítica localizada en regiones centrotemporales de hemisferio cerebral izquierdo.
Medulograma
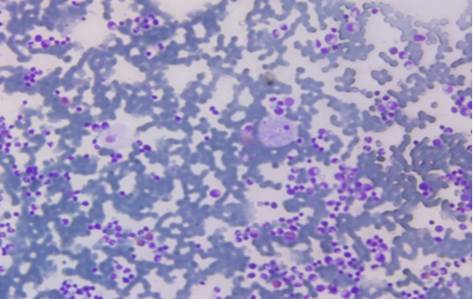
Celularidad de la médula ósea aumentada +++. Sistema megacariopoyético hiperplásico con algunas células dismórficas. Sistema eritropoyético íntegro. Sistema granulopoyético íntegro. Se observaron abundantes células de gran tamaño con uno o dos nucléolos situados excéntricamente con un citoplasma abundante de aspecto espumoso (Fig.). Las conclusiones del medulogama expresan que la paciente tiene: médula hipercelular con integridad de los tres sistemas, células compatibles con Niemann Pick.
Análisis molecular
En el gen NPC 1 se identificaron dos mutaciones en heterocigosidad (genotipo (pR1059X) + (PG904E), la primera descrita previamente como asociadas a enfermedad de Nieman Pick tipo C y la segunda identificada como variante patológica. Muestra compatible con enfermedad de Niemann-Pick tipo C. Centro de referencia: Instituto de Investigación Sanitaria Aragonés. Instituto Aragonés de Ciencias de la Salud. España.
Tratamiento
Evolutivamente la paciente recibió diversos fármacos para su epilepsia, inicialmente con carbamazepina, luego valproato en monoterapia y combinado con clobazán y finalmente la combinación de valproato y lamotrigina. La confirmación del diagnóstico de NP-C llevó a iniciar tratamiento con miglustad a dosis de 100 mg dos veces al día, ya que dosis mayores le produjeron diarreas sin resultados favorables.
DISCUSIÓN
La enfermedad de Niemann-Pick tipo C es una enfermedad neurodegenerativa y neurovisceral lisosomal de curso fatal. Se debe a una deficiencia lisosomal del transporte del colesterol no esterificado. El colesterol libre se acumula en los lisosomas y causa su disfunción. Las mutaciones del gen del NP-C1 son responsables del 95 % de los casos y las mutaciones del NP-C2 del resto de los pacientes.5,6) La presentación clínica frecuentemente no específica y heterogénea del NP-C conlleva a que el diagnóstico pueda retrasarse por varios años.4,8) Se ha publicado8 que el tiempo entre el comienzo de las manifestaciones neurológicas y el diagnóstico ha llegado a ser de 5,25 años, en esta forma infantil tardía del NP-C. La paciente que se presenta debutó con síntomas y signos neurológicos de curso progresivo e inespecífico durante varios años. Evolutivamente al constatarse la esplenomegalia, los eventos de cataplejías gelásticas y la parálisis de la mirada vertical, la sospecha clínica de que se trataba de la enfermedad de Niemann-Pick tipo C resultó alta. La cataplejía gelástica y la parálisis supranuclear conjugada de la mirada vertical son dos signos cardinales del NP-C.
El deterioro neurológico progresivo y la esplenomegalia orientaron a una enfermedad neurovisceral lisosomal. El medulograma con presencia de células espumosas así como el estudio genético resultaron compatibles con esta enfermedad, a pesar de no disponerse de las técnicas de tinción de flipin o de los hallazgos de anormalidades de la esterificación del colesterol.8 La paciente se puso bajo régimen terapéutico con miglustat, que es un inhibidor de la enzima glucosilceramida sintetasa, responsable para el primer paso de la síntesis de la glucosilceramida y de otros glicolٕípidos. El miglustad ha sido usado para prevenir la acumulación de glucosilceramida en pacientes con formas moderadas a severas de Gaucher tipo 1 y del Nieman Pick tipo C. En esta paciente no se observaron resultados alentadores con este fármaco, quizá debido a lo ya avanzado de la enfermedad.14,15,16,17,18,19
Se concluye que el deterioro neurológico progresivo, la cataplejía gelástica, paresia de la mirada vertical y la esplenomegalia, unido a los resultados del medulograma y el estudio genético permitieron el diagnóstico de esta entidad.

