










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las epilepsias focales idiopáticas (EFI), hoy nombradas focales autolimitadas, suelen tener una excelente evolución. Ellas constituyen un espectro de síndromes epilépticos sin lesión cerebral estructural subyacente, con manifestaciones clínicas y electroencefalográficas específicas. Presentan una etiología presumiblemente genética y son usualmente edad dependiente. Excepcionalmente, los pacientes afectados por estas epilepsias tienen evoluciones atípicas, que constituyen un reto diagnóstico y terapéutico.1
La regresión en niños con epilepsia puede involucrar la pérdida de habilidades cognitivas, el fallo en el progreso o un enlentecimiento de la trayectoria del desarrollo. Pocas crisis epilépticas no llevan a una regresión. Las descargas de puntas individuales en el electroencefalograma (EEG) están asociadas con alteraciones cognitivas transitorias. Cuando dichas descargas se hacen continuas, pueden provocar una regresión cognitiva del paciente. Afortunadamente, la gran mayoría de los niños con epilepsia no tienen regresión.2
Con este trabajo perseguimos el objetivo de ilustrar la evolución atípica de la epilepsia focal idiopática tipo Panayiotopoulos.
PRESENTACIÓN DEL CASO
Adolescente masculino de 13 años, presentó su primera crisis epiléptica a los 5 años de edad, mientras dormía la siesta la madre lo notó con los ojos abiertos, desviados hacia la izquierda, movía un brazo, tenía abundante salivación y presentó un vómito. Este episodio tuvo una duración aproximada de un minuto. El examen físico general y neurológico era normal, con antecedentes personales de buen aprendizaje y neurodesarrollo. Un EEG de vigilia realizado en días posteriores, mostró actividad de base normal y actividad epileptiforme interictal focal de punta onda frontocentral izquierda, que apoyó el diagnóstico de EFI tipo Panayiotopoulos, y se decidió no iniciar tratamiento con fármacos antiepilépticos (FAE).
En los siguientes tres años el niño presentó tres crisis adicionales, la última fue la más prolongada, con alteración de la conciencia, que requirió ingreso hospitalario y se decidió iniciar tratamiento con carbamazepina. En las semanas posteriores al inicio del tratamiento farmacológico, la familia y la maestra lo notaron más intranquilo, con dificultad para concentrarse y en el aprendizaje. Un EEG de sueño demostró descargas epileptiformes interictales de punta-ondas continuas durante el sueño lento (POCSL) (Fig. 1).
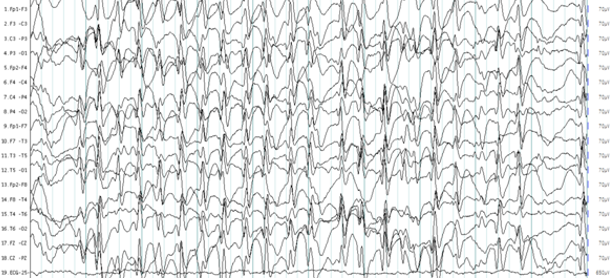
Fig. 1- EEG interictal en estado de sueño natural fases II y III de sueño no REM. Se aprecia actividad epileptiforme de punta-onda generalizada continua.
Se decidió suspender la carbamazepina e iniciar tratamiento con clobazam. En los meses posteriores mejoró la hiperactividad, pero mantuvo las dificultades en la atención y en el aprendizaje. Un estudio de resonancia magnética de cráneo no mostró alteraciones estructurales encefálicas. En EEGs evolutivos mantuvo las POCSL. Se adicionó valproato de magnesio al tratamiento y después de dos años se alcanzó la normalidad en el estudio EEG de sueño (Fig. 2).

Fig. 2 - EEG interictal evolutivo, en estado de sueño natural fase II de sueño no REM. Se aprecia la desaparición de la actividad de punta-onda generalizada continua.
Lenta y gradualmente fueron retirados los FAE, sin recurrencia de las crisis epilépticas. Evolutivamente el paciente ha mantenido las dificultades en el aprendizaje, pero ha mejorado notablemente su hiperactividad.
DISCUSIÓN
Las EFI más importantes y frecuentes son la epilepsia Rolándica (ER) y el síndrome de Panayiotopoulos (SP).3 En el caso presentado, al debut de la enfermedad, existió la interrogante en clasificar la epilepsia como ER o SP. Nos inclinamos por el SP, por la presencia de vómito ictal, y se decidió no iniciar tratamiento con FAE. Casi el 10 % de los niños con SP desarrollan crisis rolándicas puras al mismo tiempo o a edades posteriores.4 El paciente tuvo pocas crisis en un período de tres años, pero tras una crisis prolongada, se decidió iniciar tratamiento con carbamazepina, decisión que hoy en día se debate mucho, no solo el hecho de iniciar tratamiento, sino cuál fármaco utilizar.
El concepto de evolución atípica se refiere a la presencia de alteraciones neuropsicológicas severas que pueden hacerse permanentes. Se reconocen las siguientes formas: la epilepsia focal benigna atípica (EFBA), el estatus de ER, el síndrome de Landau-Kleffner (SLK) y el síndrome POCSL.1
Pacientes con dos tipos de EFI (ej., ER y SP) pueden tener una evolución atípica.5 Se ha publicado acerca de un paciente que tuvo ER y SP, con una evolución atípica, que presentó una mutación en el gen GRIN2A.6Yoshinaga y otros,7 estudiaron 5 niños que habían experimentado crisis epilépticas con manifestaciones clínicas características tanto de ER como SP, crisis rolándicas y eméticas. Ellos encontraron que todos tenían puntas rolándicas cuando tuvieron crisis rolándicas, y puntas occipitales o multifocales cuando tuvieron crisis de vómitos.
Es bien conocido que la ER, y el SP, pueden raramente evolucionar a síndromes más severos con déficits neuropsicológicos y del comportamiento. Los mecanismos fisiopatológicos que provocan estas alteraciones cognitivas no se conocen bien. La actividad EEG anormal se produce probablemente por una activación del sistema retículo-tálamo-cortical con una sincronización secundaria bilateral a través del cuerpo calloso, lo que favorece una activación de la actividad epileptiforme durante el sueño.8
Como la duración de la POCSL y la localización del foco interictal influye en el grado y tipo de disfunción cognitiva, es probable que la actividad epiléptica que ocurre durante el sueño cause los síntomas clínicos típicos por interferencia de las funciones fisiológicas relacionadas con el sueño, y posiblemente los procesos de neuroplasticidad que median las funciones corticales superiores, como son el aprendizaje y la consolidación de la memoria.8
Las manifestaciones electro-clínicas atípicas (fundamentalmente anomalías EEG y edad de debut temprana) deben ser considerados factores de riesgo para evoluciones atípicas.9,10 No hay acuerdo en cuanto al tratamiento óptimo de estas condiciones.11
Cuando existen factores de riesgo de evolución atípica, la recomendación es evitar los FAE clásicos (fenobarbital, fenitoína y carbamazepina) y algunos de los nuevos (oxcarbazepina, lamotrigina, topiramato y levetiracetam)12,13,14 y comenzar tratamiento con etosuximida, benzodiacepinas, o sultiamo. En los casos refractarios, se pueden considerar el uso de esteroides.1
El caso presentado constituye un ejemplo infrecuente de un paciente con una EFI con evolución atípica a síndrome de POCSL, probablemente inducido por la carbamazepina, con cuadro clínico-electroencefalográfico de más de dos años de duración, con mejoría favorecida por el tratamiento finalmente empleado, la evolución natural del síndrome o el efecto de ambos. Sirva este ejemplo para el debate científico de las causas que determinan estas evoluciones de las EFI y la forma de prevenirlas y tratarlas.

