










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El síndrome de Proteus (SP) es un raro síndrome hamartoso congénito con manifestaciones neuroectodérmicas. La rareza del SP contribuye a la confusión y controversia respecto al diagnóstico, ya que no todos los pacientes tienen igual grado de severidad y manifestaciones clínicas. El SP cursa con crecimiento excesivo, exagerado, asimétrico y desproporcionado de cabeza y extremidades; nevos de tejido conectivo, adiposidad disregulada de los tejidos, malformaciones vasculares, macrocefalia y/o manifestaciones neurológicas u oftálmicas.1,2,3
Se describió por Cohen y Hayden en 1979 como una entidad clínica distinta, pero fue solo en 1983 cuando Wiedeman dio su nombre, por un legendario héroe griego transformado por los dioses en un demonio inmortal del mar descrito en la Odisea de Homero. El caso más temprano fue informado por Joshep Merrick en el siglo xix, el llamado ¨ hombre elefante.1,4,5)
El SP podría estar asociado con el mosaicismo con activación somática, mutación en el gen AKT1, ubicado en cromosoma 14q32.3. Su prevalencia va desde 1:1 000 000 a 1: 10 000 000 de habitantes, mas menos de 100 a 500 casos se han publicado en la literatura en países desarrollados y en Cuba se notificó un caso en año 2015.1,5,6,7
El objetivo de este trabajo es presentar un caso clínico donde combinan dismorfias faciales, crecimiento excesivo de una hemicara, macrocráneo y manifestaciones neurológicas.
PRESENTACIÓN DEL CASO
Lactante de 10 meses. Femenina, primogénita. Hija de padres jóvenes adolescentes menores de 20 años, no consanguíneos. Embarazo no planificado o deseado, captación y atención médica tardía. Ambiente familiar disfuncional, hacinamiento, consumo de marihuana, alcohol. La madre y hermana mayor de la joven madre también tuvieron embarazos en la adolescencia; el nivel intelectual de esta madre es bajo, no terminó la secundaria.
Embarazo de 42 semanas, riesgo materno por fumadora, consumo de marihuana, bebidas alcohólicas y anemia. Parto eutócico, peso adecuado para la edad gestacional de 2900 g, perímetro cefálico (PC) al nacer de 37 cm, APGAR 9/9, sin complicaciones perinatales, estadía hospitalaria de 5 días y enviada a la consulta de genética por asimetría facial, hemihipertrofia de la hemicara derecha. Recibe lactancia materna exclusiva.
Es traída por su médico de familia a los 2½ meses, porque desde el primer mes de vida refiere movimientos de ¨contracción axial de miembros superiores e inferiores¨, que fue en incremento en frecuencia y duración, acompañado de desviación ocular hacia arriba, rubicundez seguida de irritabilidad. Durante el interrogatorio se constató e interpretó estos movimientos como espasmos infantiles (EI) en flexión, de duración variable entre 20 a 25 minutos, salvas de 40 a 60 EI, más frecuente en periodos de transición de sueño a vigilia o viceversa, hasta 245 en las primeras 24 horas de ingreso hospitalario.
Al examen físico inicial se constata: dolicocefalia, con PC de 45,5 cm> 97 percentil, cara alargada, hipertelorismo, epicanto, discreta micrognatia, asimetría facial con hemihipertrofia de hemicara derecha, boca entreabierta, comisura labial descendida y nevó hiperpigmentado, verrugoso en la región parietal ipsilateral con hoyuelos en las articulaciones de codos y rodillas Se obtuvo el consentimiento de la madre para publicar esta foto (Fig. 1).
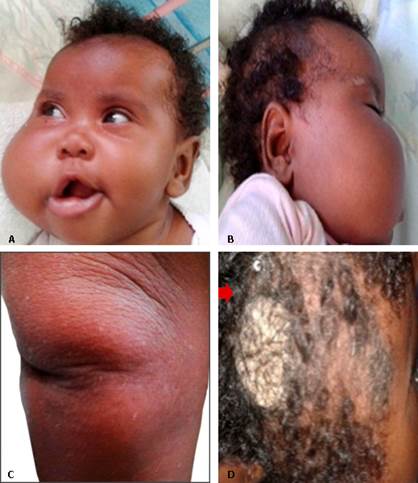
Fig. 1 A y B: hemihipertrofia de hemicara derecha, boca entreabierta y comisura labial derecha descendida. C: hoyuelo en la articulación de la rodilla. D: nevó hiperpigmentado, y verrugoso en la región parietal derecha.
Examen neurológico: estrabismo y movimientos oculares erráticos, maniobra de tracción vertical con hipotonía axial y discreto aumento del tono muscular en miembros inferiores con reflejos osteotendinosos (ROT) exaltado, sin clonus o Babinski. Fondo de ojo con papilas pálidas y grandes. El desarrollo psicomotor (DPM) estaba comprometido con escaso seguimiento visual y no sonrisa social. Se constata en el electroencefalograma (EEG), Hipsarritmia periódica y simétrica durante el sueño que hace el diagnóstico electro-clínico de síndrome de West (SW) (Fig. 2A)
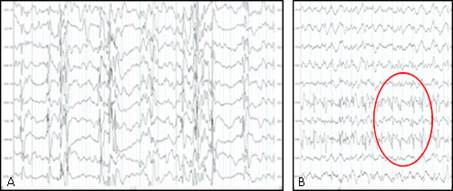
Fig. 2 EEG en vigilia. Sistema internacional 10-20. A: hipsarritmia simétrica y periódica en vigilia. B: actividad lenta tipo delta en vigilia con ojos abiertos y trazado focal centro-parietal derecha y ondas trifásicas. (EEG realizado en el departamento Neurofisiología del HPCH).
Analítica sanguínea con anemia de 10,5 g/L, microcita e hipocrómica; hierro (Fe) sérico/5 UI, TGP, TGO, CPK, LDH, amoniaco e ionograma/gasometría, dentro de límites normales.
Biopsia de piel: confirmó un nevó epitelial hiperpigmentado verrucoso inflamatorio lineal (Fig. 1D).
Ultrasonido (US) de partes blandas/ doppler: aumento de volumen del tejido celular subcutáneo (TCS) en hemicara derecha y región parietal, con doppler negativo a expensas de tejido graso y musculo. US transcraneal: disminución de las circunvoluciones cerebrales y aumento de volumen del hemisferio derecho. US abdominal y torácico: no alteraciones en órganos intrabdominales. Ecocardiograma: normal.
Sorvey óseo: engrosamiento o hiperostosis del hueso mandibular y maxilar derecho, con aumento discreto de la órbita derecha. No alteraciones en el resto de huesos largos y buen desarrollo de núcleos de osificación (Fig. 3).
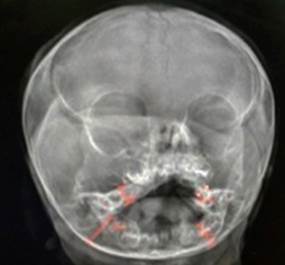
Fig. 3 Radiografía de cráneo. Vista antero posterior, con aumento del diámetro de la órbita, mandíbula y maxilar derecho. (Cortesía y Servicio de Radiología del HPCH).
Tomografía axial computarizada (TAC) de cráneo simple: cortes de 3 a 5 mm con aumento de volumen de tejido graso de la hemicara derecha capsulado, no calcificaciones y disminución de los surcos cerebrales, aumento de tamaño de hemisferio derecho a expensas de región temporal, parietal y occipital, con imagen hipodensas en su interior y dilatación del ventrículo derecho.
Resonancia magnética nuclear (RMN) de cráneo/0,35t. Se confirma hipertrofia de TCS en hemicara derecha, sin vasos en su interior. Aumento de tamaño de la órbita, mandíbula y maxilar derecho. Cráneo, con signos de hemimegancefalia derecha a predominio temporo, parieto-occipital y disminución de las circunvoluciones bilaterales, hipoplasia del cuerpo calloso. Imágenes en t1, flair hiperintensa y t2 hipointensa sugestiva de displasia cortical occipital derecha o lesión hamartomatosas (Fig. 4).

Fig. 4 Imágenes de ersonancia magnética nuclear IRMN) de 0.35t, corte sagital. A: secuencia t2, aumentodel TCS en hemicara derecha. B: secuencia t1, hemimegancefalia (HME) lobar temporo-parieto-occipital derecha e hiperintensidad. C: secuencia t2, hemimegancefalia parieto-occipital derecha con hipointensidad y engrosamiento cortical occipital. D: secuencia flair, hemimegancefalia parieto-occipital derecha e hiperintesidad cortical y de sustancia blanca.(RMN cráneo realizada Hospital Juan Manuel Márquez).
Por criterios clínicos en valoración conjunta con servicio de dermatología y genética clínica se diagnosticó síndrome de Proteus y comorbilidad con retraso del DPM y síndrome de West (SW).
Estuvo hospitalizada 28 días, por la frecuencia e intensidad de los EI, recibió tratamiento combinado de hormona adenocorticotropica sintética (ACTH) a dosis en días alternos durante 2 semanas, y dosis única semanal durante 1 semana (2 décimas intramuscular [IM]/1mg= 1 mL) con hipertensión arterial transitoria (HTA) TA >97 percentil para su edad (95/60) sin criterios para retirar la medicación a la 4ta. dosis, combinada con VGB hasta dosis de 1 250 mg día (150 mg/kg /día) en dos subdosis. Recibió tratamiento farmacológico con captopril a 1mg/kg/día en 2 subdosis durante 2 meses con resolución de la HTA transitoria, evaluada en conjunto con cardiopediatras.
A los 28 días se logró control total de los EI y se realizó EEG evolutivo con actividad lenta y desorganizada en hemisferio izquierdo y trazado focal y ondas trifásicas en región centro-parietal derecha (Fig. 2B). Al alta hospitalaria, el seguimiento es cada 15 días, solo tratamiento con VGB, sin notificar por familiares y médico de familia la recaída de EI. Evolutivamente en seguimiento en consulta mensual de neuropediatría, después del control de los EI ha logrado hitos del DPM acorde su edad. A los 10 meses, en la esfera motora se sienta sola, se mantiene sentada, rola y logra la bipedestación con apoyo, motor fino hace intercambio de objeto de una mano a la otra, con pinza digital, lenguaje con monosílabos de ta-ta, sonríe, obedece al llamado por su nombre. Al examen físico neurológico, macrocráneo de 48, 5 cm> 97 percentil con discreta hipertonía en miembros inferiores con ROT exaltados, sin clonus y Babinski. No hay evidencia de sobrecrecimiento de otras partes del cuerpo.
El manejo también incluye apoyo por trabajadores sociales, equipo de salud de atención primaria con visitas diarias al hogar para supervisar la administración correcta de la medicación y brindan a la familia el apoyo necesario en contacto estrecho con nuestra institución. Asiste de manera periódica a un área de estimulación temprana con fisiatras especializados en el seguimiento de niños con retraso en el DPM.
DISCUSIÓN
El SP es un raro síndrome hamartoso y de desorden genético, que produce sobrecrecimiento en varios tejidos (epidérmico, conectivo, óseo, adiposo y endotelial) durante la embriogénesis, por lo que las manifestaciones clínicas suelen ser evidentes desde el nacimiento o en los primeros años de vida. Constituye una enfermedad severa y con carácter progresivo por las deformidades o desfiguraciones, limitaciones físicas, estigmas sociales y repercusión en la calidad de vida.1,3,5
El diagnostico se establece por las características individuales y atendiendo al mosaicismo de la distribución de las lesiones, ocurrencia esporádica y curso progresivo (criterios generales). Nos basamos también en los hallazgos clínicos, radiológico y evolutivos.1,3,5 En nuestro caso particular, es significativa la relación existente con diferentes aspectos: hija de padres adolescentes, primogénita, exposición materna a consumo de alcohol, marihuana y tabaco, embarazo de riesgo social, captación tardía, embarazo no planificado y no deseado. De los casos publicados en la literatura hasta entonces no hay evidencia de algunas de estas relaciones.1,2,5,6,8
Atendiendo a las manifestaciones clínicas que fueron evidentes desde el nacimiento y curso de los primeros cinco meses, establecimos el diagnostico partir del trabajo de Hotamisligil7) en 1990, quien fue muy estricto y dio una puntación a cada sintomatología de las cuales el caso presentado tiene: hemipertrofia cerebral y facial derecha (5 puntos), lipomas y tumor subcutáneo en cara (4 puntos), el nevó epidérmico (3 puntos), la macrocefalia y prominencia ósea con hiperostosis del maxilar y mandíbula derecha (2,5 puntos), para un total de 14,5 puntos y el diagnostico se establecía si el paciente sumaba 13,0 puntos o más, similar al publicado por Pérez-Salomón y otros, con más de 13 puntos.5
De manera contemporánea para esclarecer todos estos criterios, en marzo de 1998, se convocó la primera conferencia nacional del SP para padres y familiares (National Institutes of Health de Bethesda, Maryland, EE. UU.), donde se establecieron criterios diagnósticos,3,8) basado en los criterios genérales y criterios específicos que se dividen en 3 categorías, A, B y C. Siendo necesario los criterios generales, la categoría A o 2 criterios de la categoría B o 3 de la C.3,8 La paciente presentada cumple los criterios generales, 2 criterios de la categoría B (nevos epidérmicos en el cuero cabelludo, hiperostosis del hueso maxilar y mandibular derecho) y 2 criterios de la categoría C (tejido adiposo disregulado en la hemicara derecha y fenotipo facial dado por macrocráneo, dolicocefalia, cara alargada, desviación de la comisura labial hacia abajo y boca entreabierta), la HME es ipsilateral a la hemihipertrofia facial; por lo que se podría concluir que padece de un SP.
A pesar de que el crecimiento desproporcionado y asimétrico de las extremidades o las malformaciones vasculares y linfáticas son signos clínicos frecuentes descritos en publicaciones de casos clínicos y son criterios específicos a considerar en el diagnóstico,1,2,3,4,5) no fueron hallados en nuestro caso y deberían observase en su evolución clínica. Razón que justifica su seguimiento según las normas de Biesecker,3) con fotografías y radiografías evolutivas u otros estudios imagenológico ante la sospecha de sobrecrecimiento o tumoraciones.
El diagnóstico diferencial se establece con las siguientes entidades: neurofibromatosis, lipomatosis encefalocraneocútanea, síndrome de Klippel-Trenaunay, síndrome de Schimmelpecinning-Feuerstein-Mims, síndrome de Maffuci, Bannayan-Riley-Ruvalcaba, Parkes-Weber y enfermedad de Ollier.1,2,3,4,5,6,7,8,9
Aunque la asociación de SP y malformaciones del sistema nervioso central (SNC) son poco frecuentes, se describen: HME, síndrome de Dandy-Walker, hipoplasia de cuerpo calloso, alteraciones del desarrollo de la certeza cerebral (heteropías, complejo agiria/paquigiria, polimicrogirias), de la sustancia blanca, calcificaciones periventiriculares y atrofia cerebral, que justifican de encontrarse en la mayoría de los casos su comorbilidad con epilepsia y alteraciones del DPM o signos clínicos al examen físico neurológico. En nuestro caso fue una manifestación clínica la asociación con el Síndrome de West y hallazgo radiológico relevante que motivó el diagnóstico clínico. Por los estudios imagenológico se confirmó en nuestra paciente, HME derecha, hipoplasia de cuerpo calloso, alteraciones de la sustancia blanca sugestiva de displasia cortical o lesión hamartomatosas en la región occipital, a pesar de no contar con imágenes de gran resolutividad.
La HME es un trastorno hamartomatoso no frecuente, debido a una alteración en la proliferación neuronal y de células gliales, dicha alteración, produce elongación de un hemisferio cerebral, acompañado de disgenesia y dismorfia celular, incluye circunvoluciones anormales y se encuentran zonas de paquigiria, polimicrogiria y lisencefalia.10,11,12,13,14) Conforme a la clasificación de malformaciones del desarrollo de la corteza cerebral según Barkovich y otros,12 pertenece a la categoría 1.C, que hace referencia a las disgenesias corticales con anomalías en la proliferación celular y asociada a síndromes, entre ellos el SP 11,12, coincidiendo en nuestro caso y el descrito por Olmos- López y otros,13 en un paciente HME en el contexto de síndrome hamartoso con SW por la triada de EI, hipsarritmia y retraso en el DPM.14) Es significativo señalar que el inicio precoz de los EI antes de los 3 meses, cuando el pico de incidencia u ocurrencia mayor de los EI es de manera habitual entre 4 a 8 meses, podría considerarse por sí solo o relacionado con la lesión estructural, como signos de mal pronóstico y son responsables en la mayoría de los casos del fracaso terapéutico y compromiso severo del DPM.14
La liga internacional de la epilepsia (ILAE) (15 en su nueva propuesta de clasificación considera la HME dentro de las causas estructurales como etiología y la relaciona con encefalopatías epilépticas de difícil control de las crisis y fármaco resistentes. En nuestro caso particular, se resalta el control de los EI, la mejoría del EEG con tratamiento combinado de ACTH y VGB según recomendaciones actuales16 y mejoría al controlar los EI y la hipsarritmia en el DPM; pero por su alteración estructural, el futuro es incierto y el seguimiento clínico debe ser minucioso, estricto y dirigido, interdisciplinario y de manera periódica.
La evolución y el pronóstico de pacientes con SP dependen del grado de afectación clínica, asociación con epilepsia, discapacidad intelectual o motora, malformaciones vasculares graves y pulmonares o tumoraciones malignas. Se ha relacionado alguna de ellas (malformaciones vasculares, pulmonares y tumoraciones) como causa de fallecimiento.1,5
Podemos concluir que el síndrome de Proteus se caracteriza por crecimiento exagerado en varios tejidos (epidérmico, conectivo, óseo, adiposo y endotelial) durante la embriogénesis, por lo que las manifestaciones clínicas suelen ser evidentes desde el nacimiento o en los primeros años de vida, se relaciona con un grupo de casos con malformaciones del sistema nervioso central y síndrome de West.

