










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El síndrome de Bland, White y Garland es un problema de salud actual con ribetes poco conocidos que precisan esclarecimiento.1 Su carácter controversial permite la asignación de nomenclatura variada; es conocida también como ALCAPA por sus siglas en inglés (anomalous origin of leftc oronary artery from pulmonary artery). En 1885 Brooks hace el primer hallazgo en dos especímenes.2 y las denomina “Origen anómalo de la arteria coronaria izquierda a partir de la arteria pulmonar.” En 1933 Bland, White y Garland describen el primer caso clínico, en cuyo honor se le comenzó a llamar “Síndrome de Bland-White-Garland”.3
La caracterización de esta entidad se configura en base a la sumatoria integrada por las manifestaciones clínicas detectadas en el paciente aquejado, las alteraciones eléctricas documentadas, los hallazgos radiológicos identificados, la historia natural y el pronóstico desfavorable sin la aplicación del tratamiento conveniente;4) se presenta de forma variada en correspondencia al momento de presentación relacionada con los cambios fisiopatológicos que acompañan al feto, al recién nacido y al lactante.5 Es una cardiopatía congénita poco frecuente, se presenta en 1 de cada 300 000 recién nacidos vivos, dato epidemiológico que proviene desde la era preecocardiográfica, letal en su historia natural con 90 % de mortalidad al año de vida.6,7,8,9
La tipificación de la dolencia reviste carácter dual al integrar modalidades catalogadas como infantil o adulta.10 En la variedad infantil se presenta con insuficiencia cardiaca. El telecardiograma muestra cardiomegalia y congestión en ambos campos pulmonares y el electrocardiograma signos de isquemia miocárdica, mientras que en la variedad adulta puede cursar de forma asintomática o presentarse como muerte súbita en el 85 % de pacientes jóvenes.11,12
Hay niños que padecen la variedad adulta del síndrome, sujetos enmarcados bajo la fachada clínica de otras entidades nosológicas o que cursan de manera silente.13 Estas situaciones provocan que resulte difícil el registro epidemiológico y conceden potencial para que la dolencia se catalogue como una de las entidades simuladoras existentes aún en la medicina moderna. Por lo que podemos alegar que los criterios epidemiológicos expuestos en la literatura están sesgados. El método clínico, que persiste como arma fundamental de sospecha diagnóstica en estos tiempos de precisión tecnológica,14,15 presenta limitaciones para contribuir al logro del adecuado índice de detección de la enfermedad, la ecocardiografía basada en la observación meticulosa y metódica pudiera ser la solución a todo lo expuesto.16,17
El objetivo de este trabajo es describir las características del diagnóstico del síndrome de Bland, White y Garland.
Métodos
Se realizó un estudio descriptivo, transversal en la totalidad de los pacientes de ambos sexos con diagnóstico confirmado de la presencia del síndrome de Bland, White y Garland que fueron pesquisados en su localidad de origen y remitidos al Cardiocentro Pediátrico “William Soler” por intermedio de la Red Cardiopediátrica Nacional, en el período de marzo de 2005 hasta noviembre del 2015.
El Cardiocentro Pediátrico “William Soler” es el centro de referencia nacional de tercer nivel, donde se realizan cirugías cardiovasculares y se ofrece cuidado intensivo posoperatorio, lo cual permitió estudiar a todos los pacientes diagnosticados en dicho período. Se excluyeron los pacientes aquejados por otras anomalías coronarias congénitas y los pacientes en los que no se confirmó el diagnóstico del síndrome de Bland, White y Garland.
Se diagnosticaron un total de 20 pacientes con síndrome de ALCAPA en el período comprendido entre marzo de 2005 hasta noviembre de 2015. La prevalencia estimada en este periodo se calcula alrededor de 2,9 por 300 000 nacidos vivos.
Se estudiaron las siguientes variables: edad al diagnóstico ecocardiográfico, género, tipo de intervención quirúrgica, supervivencia posquirúrgica y variantes del síndrome de Bland, White y Garland.
Se les realizó anamnesis, examen físico y pesquisa ecocardiográfica a todos los pacientes. El procesamiento estadístico de la información se realizó para el cálculo de la prevalencia del síndrome de Bland, White y Garland en correspondencia con los hallazgos de la pesquisa nacional a efectuar sobre la población susceptible previamente seleccionada y la información actualizada aportada por el anuario estadístico de salud.18) Se construyeron distribuciones de frecuencias absolutas que se expresaron en tablas de contingencia de doble entrada. El procesamiento estadístico de la información e utilizó la prueba exacta de Fisher, como indicador se empleó el porcentaje en las variables cualitativas. La validación estadística de los resultados de la investigación adoptó nivel de significación menor a 5 % (p< 0,05) para los grados de libertad previamente fijados en cada una de las circunstancias presentadas. Se estructuró una base de datos capaz de ser utilizada por la plataforma estadística libre R Project 3.0.1 (R Foundationfor Statistical Computing).
La investigación se sustentó en los principios de la ética registrados en la declaración de Helsinki para el trabajo con humanos.19
Resultados
En la tabla 1 se aprecia que de los 20 pacientes diagnosticados 9 pertenecen al grupo de la variedad tipo infantil y 11 pacientes a la variedad tipo adulto. La relación existente entre la edad al diagnóstico ecocardiográficoy variantes del síndrome de ALCAPA, demostró diferencias significativas donde todos los pacientes que integraron la variante tipo infantil tenía menos de 3 años de edad al momento del diagnóstico ecocardiográfico y sin embargo en la variante tipo adulto se diagnosticaron dos casos con menos de tres años; la mayoría de los pacientes eran mayores de tres años de edad. No se observaron diferencias entre los grupos relacionada con el género. Las técnicas quirúrgicas aplicadas a estos pacientes fueron la reimplantación coronaria a 9 pacientes, 6 de ellos a los de la variante tipo infantil y 3 en la variedad tipo adulto, y la tunelización se les realizó a 10 de los cuales 8 pacientes integraron la variedad tipo adulto, no hubo diferencias entre ambas variantes del síndrome en cuanto al tipo de intervención quirúrgica aplicada, como muestra la tabla 1.
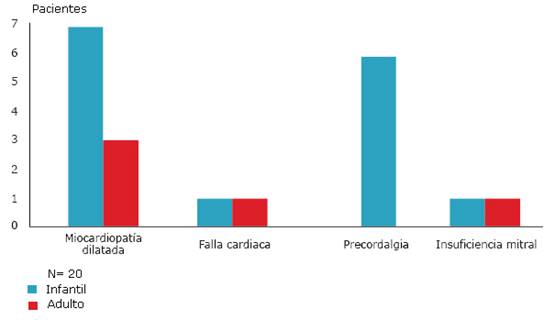
En la figura 1 se puede apreciar la expresividad clínica con la que se presentaron los pacientes estudiados, (miocardiopatía dilatada, falla cardiaca, insuficiencia mitral y precordialgia), en la variedad tipo infantil prevaleció la miocardiopatía dilatada y en la variedad tipo adulto predominó la precordialgia. La insuficiencia mitral y la falla cardiaca se manifestaron con igual periodicidad en ambas variantes de la enfermedad.
Tabla 1 Sindrome de ALCAPA.Valoración comparativa por la edad al diagnóstico ecocardiográfico, el género y el tipo de intervención quirúrgica practicada

*Paciente no intervenido; **prueba exacta de Fisher.
La tabla 2 podemos observar los pacientes pertenecientes a la variante infantil sobrevivieron el 62,5 %, y los de la variante adulta el 100 %, independientemente de la técnica quirúrgica aplicada para una supervivencia posquirúrgica en el síndrome de ALCAPA s general de 84 % (p= 0,05).

Discusión
Los estudios publicados previamente se limitan a reporte o series de casos debido a la baja incidencia descrita de la enfermedad.4,7,9,11,13,20,21,22
La prevalencia perteneciente a la primera década del siglo xxi en Cuba es mayor que lo registrado hasta el momento (1 de cada 300 000 recién nacidos vivos).20) En el presente estudio se demuestra la variabilidad en su presentación y la alta supervivencia con el tratamiento quirúrgico, tal y como se describe en la literatura.17,21
Es esencial el diagnóstico temprano del origen anómalo de la arteria coronaria izquierda a partir del tronco de la arteria pulmonar para iniciar lo antes posible su tratamiento y con ello mejorar su supervivencia y pronóstico.22) En su historia natural solo el 15 % desarrollan circulación colateral teniendo una expectativa de vida de 30-40 años.23
La indicación quirúrgica en el paciente con diagnóstico confirmado de síndrome de Bland, White y Garland es clara. Se han descrito diversas técnicas quirúrgicas con el fin de interrumpir el robo de flujo coronario a la arteria pulmonar o para ofrecer flujo sistémico a la coronaria izquierda.24,25) Existe consenso respecto a la superioridad del restablecimiento del sistema de doble irrigación coronaria sobre la ligadura de la coronaria anómala, debido a que se relacionan con alta mortalidad,26,27) y es en la actualidad la técnica de elección el reimplante directo que exhibe excelentes resultados en diversos centros hospitalarios,28,29) pero hay un grupo de casos que por sus características anatómicas, resulta difícil la implantación de la coronaria en la aorta y debe aplicarse entonces la técnica de Takeuchi.30
Las técnicas quirúrgicas utilizadas en el presente estudio fueron el reimplante coronario: transferencia del botón coronariano hacia la arteria aorta y la técnica de Takeuchi que consiste en la tunelización del ostium de la coronaria anómala en el tronco de arteria pulmonar hasta la aorta a través de una ventana aortopulmonar quirúrgica.
El acometimiento quirúrgico ante el síndrome de Bland, White y Garland es una realidad tangible pero la caracterización clínica abigarrada, oculta o silente de la dolencia limita la sospecha diagnóstica sobre bases tradicionales, por lo que se bosqueja el mencionado síndrome como un problema de salud actual con ribetes poco conocidos que precisan esclarecimiento. El carácter silente ocasional, el mimetismo clínico y las diversas formas de expresión presentes de acuerdo con la edad de los individuos aquejados inducen a la concepción de nuevas estrategias clínicas y complementarias como elementos facilitadores para la detección y el enfrentamiento exitoso de la dolencia.
Podemos concluir que el diagnóstico del síndrome de Bland, White y Garland es difícil mediante el método clínico debido a la diversidad en su presentación. La prevalencia estimada en Cuba es mayor que lo reportado en el mundo hasta la actualidad. El diagnóstico temprano y tratamiento oportuno mejora de forma significativa la supervivencia y el pronóstico de los pacientes aquejados por esta dolencia.
Se recomienda la práctica del examen ecocardiográfico ante la sospecha clínica de dicho síndrome en pacientes pertenecientes a cualquier grupo de edad y la necesaria realización de estudios multicentros dirigidos a elevar la calidad del diagnóstico no invasivo en esta enfermedad.

