










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las inmunodeficiencias primarias son enfermedades genéticas, dado por un déficit cuantitativo o cualitativo en uno o más componentes del sistema inmunológico, que trae como consecuencia infecciones recurrentes, entre otros síntomas y signos importantes.1
Estas afecciones están constituidas por más de 200 entidades. El síndrome de Job o SHIE se incluye dentro de los síndromes bien definidos con inmunodeficiencia,2) nombrado así en1966 cuando se describieron dos niñas con abscesos “fríos” recurrentes por estafilococos, neumonía y una erupción eccematoide de aparición en el período neonatal, que les recordaron un personaje bíblico del antiguo testamento que sufría de lesiones cutáneas recurrentes (Job 2:7). Años más tarde (1972), Buckley citado por Guisado y otros,2) comunica la existencia en estos pacientes de concentraciones séricas de IgE excepcionalmente elevadas y normales en el resto de las inmunoglobulinas.
En 1999 se estableció definitivamente la forma de transmisión del SHIE clásico, a través de una herencia autosómica dominante con penetrancia variable (SHIE-AD). Luego se identificó una mutación homocigota del gen de la tirosina quinasa 2 (Tyk2) como causa molecular del SHIE autosómico recesivo (SHIE-AR). Recientemente, se describió la mutación de DOCK8 (dedicator of cytokinesis 8) como otra forma de transmisión de SHIE-AR, cuya patogenia es desconocida. Guisado y otros.2,3)
El SHIE se considera un desorden por disregulación inmune, complejo, caracterizado por hiperglobulinemia E, eosinofilia, abscesos cutáneos, dermatitis eccematoide crónica, candidiasis mucocutánea e infecciones pulmonares recidivantes con desarrollo de neumatoceles y bronquiectasias; cuya herencia puede ser autosómica recesiva o dominante.3,4)
Se clasifica en: tipo I, SHIE-AD (autosómico dominante), en este los pacientes presentan anomalías en distintos órganos (tejido conectivo, esquelético y vascular; el tipo II, SHIE-AR, (autosómico recesivo) también afecta al sistema inmune. Este cursa con IgE elevada, infecciones recurrentes de piel y pulmón, susceptibilidad a infecciones virales (Molluscum contagiosum), y compromiso del sistema nervioso central, sin alteraciones musculo esqueléticas. Los pacientes con SHIE comparten rasgos comunes pero diferente compromiso clínico e inmunológico; en la mayoría de los casos se desconoce el defecto molecular.5)
Aproximadamente 25 % de los pacientes con SHIE presentan mutaciones en STAT-3, donde se observan características inmunológicas y no inmunológicas, que incluyen: fascie característica; se evidencia luego de la adolescencia, frente y barbilla prominente, puente nasal ancho, cráneo sinostosis, retención de dientes primarios, paladar ojival, hiperlaxitud, escoliosis, osteoporosis y fracturas patológicas. Estos pacientes tienen riesgo elevado de desarrollar malignidad, particularmente linfoma no hodgkiniano y aneurismas arteriales.4,5
Aquellos pacientes que presentan mutaciones en DOCK8 desarrollan una respuesta inmune defectuosa. En ellos, los linfocitos T CD3+/CD4+ y CD3+/CD8+están disminuidos, presentan un deterioro de la función proliferativa luego de la estimulación in vitro y alteración en la producción de citocinas antivirales. No presentan anormalidades esqueléticas ni dentarias y sí, complicaciones autoinmunes. Las mutaciones en DOCK8 generan un fenotipo de SHIE con menor compromiso pulmonar pero mayor expresión de síntomas cutáneos y aumento de la susceptibilidad a las infecciones virales. Las deficiencias en Tyk2 cursan con IgE con concentraciones menos elevadas o normales y tienen un fenotipo clínico muy variable. Se presenta una disfunción en la respuesta inflamatoria, con deficiencia de los mecanismos celulares de la respuesta inmune frente a agentes infecciosos y reparación tisular anómala, asociada a la incapacidad para orquestar una respuesta específica Th17.4,5
El objetivo de esta comunicación es contribuir al conocimiento de esta inmunodeficiencia primaria para su detección precoz y tratamiento oportuno.
Presentación del caso
Se describe una lactante de seis meses de edad, del sexo femenino, antecedentes patológicos familiares de alergia y diabetes mellitus tipo II. Antecedentes prenatales de infección urinaria en el 1er. trimestre. Nació a las 39,1 semanas, producto de un parto distócico por cesárea anterior. Peso de 3818 g, Apgar 9/9. Presentó un episodio de hipoglicemia al nacer, sin otra complicación perinatal. Recibió lactancia materna exclusiva hasta los 4 meses. Esquema de inmunización actualizado según su edad. No tuvo reacción posvacunal.
Desde su nacimiento ha tenido infecciones respiratorias altas y bajas recurrentes, complicadas y no complicadas (otitis media aguda, herpangina, neumonías, bronquiolitis, síndrome coqueluchoide, laringitis). A los 11 días de nacida tuvo el primer episodio de infección respiratoria alta, posteriormente presentó 8 eventos infecciosos en un período de 5 meses, de diversa etiología: bacterianos, virales y fúngicos; dados por eczemas recurrentes en cuello, axilas, región inguinal y perianal, así como lesiones micóticas en piel (candidiasis mucocutánea) y gastroenteritis, manifestado por diarreas persistentes e intolerancia a la leche de vaca.
Estos episodios requirieron múltiples ingresos y tratamiento con antimicrobianos: penicilinas, cefalosporinas de 1ra. y 3ra. generación, sulfas, fosfocina y antifúngicos. A los 5 meses de edad ingresó en la unidad de cuidados intensivos, por una sepsis grave (celulitis orbitaria preseptal de hemicara derecha. Al examen físico se constataron: signos inflamatorios agudos en párpado inferior derecho con pequeña área de fibrosis, puente nasal ancho. Leve protrusión de la frente. Eczemas en la región del cuello, ambas axilas, región vulvar y perianal, acompañado de prurito. Las manifestaciones musculoesqueléticas comúnmente descritas en esta enfermedad: cifoescoliosis y fracturas patológicas no se constataron al momento del diagnóstico. (Fig.)

Se indicaron complementarios para el perfil de sepsis, instaurándose el protocolo establecido. Se interconsultó con otras especialidades: oftalmología, dermatología, alergia e inmunología. La evolución fue tórpida, aparecieron complicaciones: neumonía, neumatocele (Fig.), extensión de la infección orbitaria, facial y repercusión sistémica de la infección. Se utilizaron múltiples antibióticos para el control de la sepsis, que se logró.
Los complementarios al ingreso demostraron una infección bacteriana severa con leucocitosis a predominio de neutrófilos; leucos: 25 X 109/L, polimorfonucleares neutrófilos: 0,88; linfocitos: 0,10; eosinófilos: 0,02; eritrosedimentación: 112 mm/s. Tras el control del proceso infeccioso, sus cifras se normalizaron. Se aisló Staphylococcus aureus en hemocultivo.
El exudado nasofaríngeo resultó positivo para S. aureus; así como el estudio microbiológico de las lesiones en piel. Se comprobó infección por Candida albicans.
El estudio citomorfológico mostró eosinofília moderada, con valores absolutos de eosinófilos de 450 X 109 [valores normales (VN): hasta 350 X 109], concentración de IgE de 49,93 UI/ml (VN: 15 UI/ml).
La evaluación de la función fagocítica detectó un marcado trastorno de la fagocitosis expresado a los 15 y 60 minutos, respectivamente: T15_ 74,00 % (VN: 22,99 a 53,95 %), T60_ 63,60 % (VN: 6,63 a 28,43 %). La cuantificación de inmunoglobulinas resultó dentro de los parámetros normales: IgG_ 5,20 (VN: 3,90-7,94 g/L), IgA_0,32 (VN: 0,01-0,59 g/L), IgM_1,3 (VN: 0,09 a 2,12 g/L), según valores de referencia para la edad. 6
En el estudio celular de las subpoblaciones linfocitarias por citometría de flujo, se constató ligero incremento de las células T CD3+/CD8+: 27,53 % (VN: 13-26 %). CD3+: 71,46 % (VN: 50-77 %), CD3+/CD4+: 36,22 % (VN: 33-58 %), CD19+: 17,82 (VN: 13-35 %), CD56+: 6,01(VN: 2-13 %). El resto de las subpoblaciones resultaron normales.7
Se realizó un fenotipo ampliado de linfocitos T y B con los resultados en la tabla 1.

Con el objetivo de evaluar los diferentes compartimientos dentro de estas importantes poblaciones linfocitarias, se realizó el fenotipo ampliado de linfocitos T y B, respectivamente. Los principales trastornos de la homeostasis encontrados en dichas subpoblaciones fueron: disminución marcada de linfocitos TCD4+ y linfocitos TCD8+ vírgenes y de memoria, respectivamente, así como de las células TCD3+ y las células B activadas (CD19+CD38+CD27+) como se muestra en la tabla 1.
Teniendo en cuenta los antecedentes descritos anteriormente, la edad de comienzo tan temprana de los síntomas, la sepsis grave con evolución tórpida, los hallazgos clínicos constatados y los criterios de Grimbacher, así como los exámenes de laboratorio, se sospechó el diagnóstico de un síndrome de hiperIgE autosómico dominante (SHIE-AD).1,2 El caso que se presenta es una de las pocas pacientes diagnosticadas en edades tan tempranas de la vida en Cuba.
Los pilares terapéuticos se fundamentaron en la orientación de medidas profilácticas para el cuidado de la piel, mucosas y de las vías respiratorias. El tratamiento medicamentoso para la estimulación/modulación del sistema inmunológico y el control de las infecciones. Se indicó factor de transferencia, (Hebertrans, del laboratorio productor del Centro de Ingeniería Genética y Biotecnología, Cuba); 1 unidad subcutánea 3 veces por semana por 8 semanas, combinado con Intacglobín, (gammaglobulina intravenosa bulbo 2,5 g/ 50 m/L, del laboratorio productor de la Empresa de Sueros y Productos Hemoderivados "Adalberto Pesant", Cuba); a dosis sustitutiva de 200 mg/kg (5 dosis); vitaminoterapia, antihistamínicos: ketotifeno; antifúngicos: ketoconazol y nistatina, para el control de los eccemas y la candidiasis mucocutánea.
Tras el tratamiento se comprobó una notable mejoría clínica; la paciente permaneció asintomática durante 6 meses. Hubo disminución de las lesiones eccematosas y abscesos en piel. Las infecciones respiratorias disminuyeron con regresión del neumatocele.
Discusión
Las características clínicas descritas en esta entidad, según porcentaje de frecuencia aproximada, se dividen en: inmunológicas: IgE> 2000 IU/ml, eczema moderado-severo (97 %), eosinofília (95 %), neumonías recurrentes (90 %), erupción en el período neonata (87 %), candidiasis mucocutánea (83 %), otitis y sinusitis recurrente (80 %), bronquiectasias, neumatoceles y abscesos (70 %); y no inmunológicas: fascies característica (85 %), lesiones hiperdensas en RNM cerebral (75 %), hiperlaxitud (70 %), retención en la dentición primaria (70 %), fracturas patológicas (65 %).8
Se aplicaron los criterios del Instituto Nacional de Salud de los Estados Unidos (NIH), denominados de Grimbacher (Tabla 2), se obtuvo un puntaje de 55 puntos, por lo que se concluyó como diagnóstico probable de SHIE- AD: ya que el puntaje total sumó más de 40 puntos, a pesar de no contar con estudios genéticos.3,8
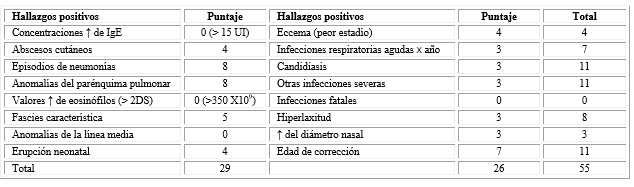
Esto sindicadores se evaluaron teniendo en cuenta que la paciente presentó las manifestaciones a los 6 meses de edad. También, se muestra un amplio rango de manifestaciones somáticas no inmu nológicas.8,9
Una característica típica del SHIE-AD es la inflamación aberrante, que lleva a la producción de abscesos fríos en la piel con una respuesta inflamatoria exagerada, principalmente pulmonar, que provocaría la formación de neumatoceles. Esto se debe a que el STAT-3 actúa como un activador de la señalización de diferentes receptores de citocinas, que incluyen IL-6, IL-10, IL-11, IL-17, IL-21, IL-22 e IL-23, entre otros. El hecho de que STAT-3 participe, tanto en vías de citocinas pro- como antinflamatorias, conduce a una desregulación que inhibe la inflamación según las circunstancias.2,10,11
Las mutaciones en STAT-3 determinan una falla en la diferenciación del patrón de respuesta Th17 y en la secreción de IL-17.2,3 Existen evidencias que una deficiencia en el linaje Th17 incrementa la susceptibilidad de la infección por S. aureus, y los efectos protectores de IL-17 son significativos en pacientes con SHIE, susceptibles a infecciones recurrentes por este patógeno. Se conoce que IL-6 e IL-23 son necesarias para el desarrollo de Th17 y activan a STAT-3. La deleción de STAT-3 se asocia, además, a la generación de osteoclastos, osteopenia de fenotipo osteoporótico y niveles más elevados de reabsorción ósea.10,11
Las células Th17 se cretan también IL-22, la cual participa en la regulación positiva de distintas células para la producción de CCL20 y β defensinas, como, por ejemplo, queratinocitos y células epi teliales pulmonares. Se ha demostrado en pacientes con síndrome de hiper IgE- AD (autosómico dominante), una reducción en la producción de esos péptidos antimicrobianos. Además, la anómala remodelación pulmonar hallada luego de infecciones o cirugías, se ha relacionado con la participación de STAT-3 en la regulación de distintas metaloproteinasas de la matriz pulmonar. (10,11 La disminución de la producción de defensinas en la piel y el pulmón puede explicar los abscesos en piel y pulmón característicos del SHIE.11
Existe una alteración en la estimulación de la producción del factor antiestafilocócico del epitelio bronquial y los queratinocitos, lo que explicaría la restricción de las infecciones por S aureus en estos órganos. (2,5 Estas neumopatías pueden ocurrir más tardíamente, particularmente en niños tratados con profilaxis antibiótica; sin embargo, en el caso que se presenta, aparecieron en etapas tempranas de la vida.10,11
En general, el S. aureus es la bacteria más frecuentemente aislada en las muestras microbiológicas de estos pacientes. Durante los episodios infecciosos también pueden aislarse: Streptococcus pneumoniae, Haemophylus influenza, enterobacterias, Pseudomona aeruginosa y gérmenes oportunistas como Pneumocystis jiroveci y Criptococcus, causantes de neumopatías e histoplasmosis. Los pacientes con SHIE presentan susceptibilidad a infecciones fúngicas que incluyen moniliasis oral y onicomicosis por C. albicans. Las infecciones forman abscesos y neumatoceles o quistes pulmonares, que pueden sobreinfectarse por Aspergillus fumigatus y Pseudomonas aeruginosa multirresistentes.2,4,12
Otro hallazgo característico de estos enfermos, es la presencia de dermatitis crónica, pruriginosa, desde las primeras semanas de vida. Sus características y distribución difieren de la dermatitis atópica, con mayor compromiso en la cara, axilas, región inguinal, periné y superficies de extensión. A diferencia de los pacientes con dermatitis atópica, en el SHIE suele faltar el antecedente familiar de atopia y las infecciones cutáneas no son solamente superficiales, sino que afectan al tejido celular subcutáneo, como se comportó en la paciente que se presenta.13
El retraso en la caída de la dentición primaria, es otro rasgo clínico que puede ayudar al diagnóstico, se observa en el 60 - 72 % de los pacientes. El retraso en la segunda dentición asociado a la erupción de los sucedáneos, puede determinar la presencia de una doble hilera de dientes.1,14 Por la edad tan precoz de aparición de los síntomas no apareció este signo clínico en el caso que se presenta.
Las anomalías óseas y del tejido conjuntivo pueden presentarse a cualquier edad. Los pacientes presentan fracturas con poco dolor y densidad ósea eventualmente normal. Sin embargo, particularmente el aplastamiento de las vértebras lumbosacras se encuentra casi exclusivamente en adultos o niños mayores y se asocian con osteoporosis. Más de 75 % de los mayores de 11 años presentan escoliosis. También puede observarse hiperextensibilidad de las articulaciones. Se considera que el defecto de señalización de IL-11, sería la causa primordial de las malformaciones óseas y dentales, no observado en la paciente.14
El estudio inmunológico de la lactante mostró concentraciones elevadas de IgE, (49,90 UI/ml) superiores a valores esperados para su edad. Existe un porcentaje de recién nacidos con SHIE que pueden presentar concentraciones normales y hasta el 20 % de los adultos llegan a normalizarlos. El aumento no es proporcional a la gravedad del cuadro y un gran porcentaje está dado por la IgE específica a S aureusy C albicans.13 Las concentraciones de IgG, IgA e IgM generalmente se encuentran normales, tal como se comportó en el caso que se presenta.
Habitualmente, el hemograma en estos enfermos no muestra alteraciones, aunque se ha descrito neutropenia relativa. Algunos pacientes muestran eosinofilia relativas de hasta 40-50 % y absolutas mayores a 2 desviaciones estándar. (6 El hemograma del caso que se presenta demostró una leucocitosis a predominio de polimorfonucleares neutrófilos en el curso de una sepsis grave, descrita anteriormente y evolutivamente estos valores se normalizaron. La producción del factor estimulador de colonias granulocitos-monocitos se ha encontrado elevada en estos pacientes, lo que explicaría la eosinofília.2,9,11 Se constataron valores altos de eosinófilos: 45 X 109/L.
La mayoría de los autores informan valores normales de linfocitos T, deficiente producción de anticuerpos específicos y disminución de linfocitos B de memoria. Este último hallazgo se corroboró en el fenotipo ampliado de linfocitos T y B, realizado. Se constató disminución de LTCD4+, LTCD8+ vírgenes y LTCD4+ memoria, así como una disminución de linfocitos T CD3+ y linfocitos B activados. Este resultado coincide con otro grupo de autores que describen una disminución de la inmunidad celular.3,5,10
A diferencia de la enfermedad granulomatosa crónica, uno de los principales diagnósticos diferenciales del SHIE; en el caso que se presenta, se comprobó un trastorno importante en la fagocitosis, descrito anteriormente. 8 Las células Th17, comprometidas con esta afección, son importantes en el reclutamiento de neutrófilos y la adhesión celular, eventos claves en la inmunidad frente a patógenos. Por otra parte, el déficit en la producción de IL-12 no favorece la activación de los macrófagos y por tanto sus potentes mecanismos microbicidas.2,5,10) Algunos autores refieren que existe deficiencia de receptores para C3b del sistema de complemento en los neutrófilos, lo que sería un importante factor promotor de la quimiotaxis y la fagocitosis.13
El enfoque terapéutico de esta enfermedad está orientado al control de los síntomas; el tratamiento oportuno de las infecciones y las complicaciones,15 resumidas en:
Cuidados dermatológicos (cremas, humectación y antisépticos)
Contraindicación de vacunación BCG
Drenajes de abscesos
Medidas para osteopenia (dieta, vitamina D y calcio)
Profilaxis antibacteriana con sulfametoxazol-trimetroprim (TMS)
Profilaxis antimicótica con fluconazol/ itraconazol
Suplemento con gammaglobulinas (si hubiera además deficiencia de anticuerpos)
Cuidados neumológicos
Las infecciones bacterianas o fúngicas diseminadas se tratan con (SMX-TMP), de elección en la prevención de las infecciones cutáneas estafilocócicas e infecciones respiratorias, el fluconazol es el agente de elección en las candidiasis mucocutánea.14,15
La infusión con gammaglobulinas intravenosa no ha demostrado ser efectiva. Sin embargo, en esta paciente resultó muy favorable, aunque se usó combinada con el factor de transferencia.2,14 Se ha utilizado con muy buenos resultados, el interferón gamma recombinante humano, los anticuerpos monoclonales rituximab (anti CD20) y omalizumab (anti IgE). El trasplante de médula ósea sigue siendo controversial y evidentemente la terapia génica es la solución definitiva en estos pacientes.8,15
Se concluye que el SHIE-AD es una inmunodeficiencia primaria poco frecuente caracterizada por altas concentraciones de IgE, eosinofilia, abscesos cutáneos, eccemas, candidiasis mucocutánea crónica e infecciones pulmonares recidivantes, neumatoceles y bronquiectasias; también se presentan alteraciones del tejido conectivo, esquelético y vascular. Se requiere de alto grado de sospecha clínica y es importante la atención precoz de las infecciones, que en general presentan una respuesta tórpida sistémica. Las alternativas terapéuticas están dirigidas a prevenir la sepsis y al control de los síntomas.

