










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El sistema nervioso se forma a partir de la capa embrionaria más externa y gruesa, el ectodermo. (Fig.1)
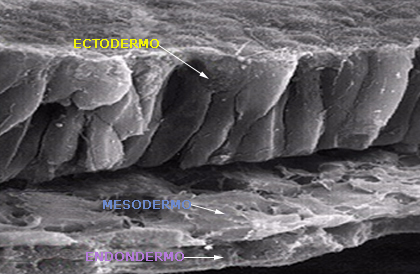
Fuente: Arslan O. Developmental aspect of the nervous system. Neuroanatomical Basis of Clinical Neurology. 2001. The Parthenon Publishing Group Inc.
Fig. 1 Microfotografía electrónica de barrido de una sección transversal de un de ratón equivalente a un embrión humano de 17 días.
Evolutivamente, a partir de la placa alar del metencefalo se origina el puente y el cerebelo. (Fig. 2)

Fuente: Arslan O. Developmental aspect of the nervous system. Neuroanatomical Basis of Clinical Neurology. 2001. The Parthenon Publishing Group Inc.
Fig. 2 Representación esquemática del metencefalo y primeros esbozos de formación del cerebelo a partir de la placa alar.
El desarrollo embrionario del cerebelo hasta su estado adulto, ha sido posible seguirlo gracias a los estudios de imágenes y poder establecer una correlación clínica más adecuada entre su desarrollo y funciones evolutivas de este órgano. (Fig. 3)

Fuente: Takahashi E, Hayashi E, Schmahmann JD. Neuroimagen. 2014;96:326-33.
Fig. 3 Vista sagital de la corteza cerebelosa. Desarrollo de la conectividad cerebelosa en el feto humano, puesto de manifiesto por tractografía de difusión angular de alta resolución, a diferentes semanas de gestación y hasta la edad adulta.
El niño sano entre los 12 y 14 meses comienza a andar y se puede constatar en ese período que avanza, amplia el plano de sustentación y se muestra inestable, expresión de la inmadurez del sistema nervioso y en especial del cerebelo, la ataxia fisiológica del niño a estas edades.
El cerebelo está localizado en la fosa posterior, por debajo del repliegue que forma la duramadre, por donde transcurre parte del drenaje venoso supratentorial a través del seno recto, la llamada tienda del cerebelo; por delante el cuarto ventrículo separa al cerebelo de la cara posterior de la protuberancia y por debajo la concha del occipital donde descansa este órgano.1.2,3,4
El objetivo de este trabajo es examinar las principales características anatómicas y funcionales del cerebelo y relacionarlas con su expresión clínica cuando enferma, así como comentar sobre su abanico de etiologías en el niño.
Métodos
Las fuentes de búsquedas fueron las bases de datos computarizadas: PubMed, Ebsco, SciELO. Se utilizaron para la búsqueda las palabras clave: cerebelo, ataxia, errores congénitos del metabolismo y ataxias, ataxias episódicas, enfermedades progresivas del sistema nervioso y ataxias; en idioma español e inglés.
Resultados
Ha sido tradicional aceptar que el cerebelo tiene una función motora, que coordina, sincroniza la actividad motora voluntaria, los movimientos oculares, regula la fonación, mantiene el equilibrio y procesa los impulsos exteroceptivos.5,6,7
En las últimas décadas unido a este “cerebelo motor” se ha ido imponiendo la certeza de que el cerebelo participa en funciones cognoscitivas, que es “un temporizador”, que ordena temporalmente las acciones para construir secuencias tanto motoras como cognitivas. Participa en la programación, planificación, ejecución y monitoreo de procesos cognitivos (solución de problemas y tareas de asociación). Participa en la memoria, aprendizaje, conducta emocional y modulación autonómica.8,9,10,11,12,13,14
El síndrome cerebeloso cognitivo afectivo es un ejemplo, expresado por alteraciones en las funciones ejecutivas, alteraciones en la memoria espacial, déficit de lenguaje y cambios de personalidad, eventos que suceden luego de la resección de tumores de fosa posterior que comprometen al cerebelo.3,4
El cerebelo se puede estudiar en su evolución filogénica y en la adquisición de funciones cada vez más complejas, desde los animales inferiores (peces, anfibios y aves) hasta los mamíferos incluido el hombre.
Para el logro de sus funciones es muy notoria la cantidad de información que llega al cerebelo desde centros vecinos y médula espinal y las que parten de él en dirección a estructuras cerebrales, que tienen la responsabilidad de planificar, organizar y ejecutar el plan motor del organismo.1,2
Entre sus principales funciones están el control del equilibrio, el control postural de los músculos que se oponen al efecto de la gravedad y sobre todo, la coordinación de la motilidad voluntaria.5,6,7
El cerebelo recepciona múltiples informaciones procedentes de los músculos, tendones, articulaciones y de otros órganos especializados como el aparato vestibular y ocular y envía información a diversas estructuras cerebrales por medio de las cuales modula la excitabilidad de estas estructuras y sus sistemas descendentes.
Es como el director de una gran orquesta, que, sin tocar algún instrumento en específico, organiza, dirige, coordina múltiples funciones que se traducen en fuerza, tiempo y secuencia.
El cerebelo enfermo impide que la persona ejecute sus funcionesy movimientos de una forma uniforme y coordinada. Puede afectarse todo el cerebelo o parte de él.5
Su forma recuerda una mariposa, con sus partes laterales (hemisferios cerebelosos) y una central (vermis). Dependiendo de la parte que resulte afectada, así serán las manifestaciones clínicas. Un ejemplo de compomiso del vermis cerebeloso es la intoxicación alcohólica, todos hemos visto a estas personas que son incapaces del control de la cabeza y del cuerpo y tienen una marcha muy peculiar.1,2
Cuando se afecta un solo hemisferio cerebeloso el paciente muestra signos localizados en un hemicuerpo, que se corresponden con el hemisferio cerebeloso enfermo del mismo lado: temblor, dismetría, hipotonía, entre otros.
Cuando se afecta el cerebelo como un todo, como ocurre en algunas enfermedades inflamatorias infecciosas o no, se producen síntomas y signos tanto vermianos como hemisféricos.
La ataxia es una de las expresiones clínicas del paciente con afección del cerebelo, pero puede resultar del compromiso de otras estructuras dentro del sistema nervioso.6,7
Taxia es sinónimo de coordinación, es el resultado de la combinación de la contracción de los músculos agonistas, sinergistas y fijadores, así como la relajación de músculos antagonistas que tiene como resultado que los movimientos sean armónicos, coordinados y mesurados.
Cuando el paciente tiene afectada esa capacidad de coordinación precisa de la actividad muscular, el paciente está atáxico.
Existen diversas afecciones del sistema nervioso que se manifiestan por ataxia, por ejemplo:
Afecciones de nervios periféricos y de los cordones posteriores de la médula espinal que impiden que lleguen a diversas estructuras cerebrales, sobre todo al cerebelo, la información propioceptiva correspondiente. (ataxia sensitiva)
Enfermedades del laberinto, nervio y vías vestibulares.
Lesiones frontales que pueden expresarse por apraxia/ataxia (ataxia frontal de Bruns)
Lesiones del cerebelo o sus vías
La afección del cerebelo produce un conjunto de síntomas y signos (síndrome cerebeloso) donde la ataxia es uno de los más prominentes.5,6,7
El clínico ante un paciente con ataxia debe precisar el sitio de la afección neurológica que la ocasiona a través de una correcta anamnesis, examen físico general y neurológico, diagnóstico topográfico, diagnóstico positivo y diferencial, diseño del protocolo de estudio y su interpretación (método clínico).
En el examen del paciente atáxico es útil determinar si hay certeza a la exploración física, del signo de Romberg (en la estación de pie, la ataxia empeora al cerrar los ojos), es clásico afirmar que el paciente cerebeloso no lo presenta y sí lo muestran tanto los que tienen afectación de la vía propioceptiva, como los que tienen comprometida la aferencia desde el aparato vestibular o sus vías.
El cerebelo puede resultar afectado por un amplio abanico de posibilidades etiológicas, genéticas o adquiridas (agenesia, atrofia, megacerebelo),15) congénitas o de aparición posnatal, malformativas o de curso heredo-degenerativo, inflamatorias infecciosas o no infecciosas, vasculares, tumorales, tóxicas, traumáticas, entre otras.
Una forma útil de orientar el proceso diagnóstico, resulta en clasificar las ataxias cerebelosas según su evolución en el tiempo, ataxias agudas, agudas recurrentes, ataxias crónicas no progresivas y las crónicas progresivas.5,6,7
Ataxias agudas
Existe un grupo de causas que pueden afectar al cerebelo en los niños de comienzo agudo y entre las más frecuentes las tóxicas, traumáticas, las inflamatorias infecciosas y no infecciosas y vasculares, entre otras.
En tiempos de la COVID-19, se ha documentado en algunos niños con esta enfermedad, un conjunto de síntomas y signos que remedan la enfermedad de Kawasaki y tanto esta como la COVID-19 pueden comprometer al sistema nervioso y afectar el cerebelo.
Ataxias agudas recurrentes o episódicas
Estas son menos frecuentes y constituyen en ocasiones un reto para el médico; varios errores congénitos del metabolismo (ECM) pueden expresarse por una ataxia recurrente episódica (AE), períodos que alternan de normalidad y de nuevo la ataxia. (deficiencia de piruvato deshidrogenasa, enfermedad de Hartnup, enfermedad de la orina con olor a jarabe de arce).16,17,18,19,20,21,22,23,24,25
Enfermedades genéticas no metabólicas, ataxias episódicas
Constituyen un grupo de enfermedades que se heredan con un patrón autosómico dominante, representadas por presentar períodos de descoordinación asociados a otros síntomas o signos, por ejemplo, mioquimias (movimientos musculares continuos). Se desencadenan sobre todo por factores ambientales como stress físicos o emocionales. Pueden presentarse desde la infancia temprana hasta la adultez. Se ha descrito más de media docena de estas ataxias recurrentes, aunque dos tipos son los mejores caracterizados y frecuentes dentro de su rareza (1 en 100 000 personas): AE tipo I con mioquimias y la tipo II, donde las mioquimias son raras.
Ante un niño que sin razón aparente se pone atáxico de forma recurrente tener presente el síndrome de Munchausen por poder, donde el cuidador le ocasiona por alguna vía este importante signo clínico al niño.
El paciente que de forma congénita o posnatal sufre una lesión cerebelosa, puede quedar con manifestaciones atáxicas de manera permanente, pero sin empeoramiento progresivo (malformaciones, traumas, infecciones, intoxicaciones).25
Sin duda, los pacientes con ataxias cerebelosas crónicas progresivas constituyen un grupo muy importante de pacientes, al que debemos diagnosticar y tratar.26,27,28,29,30,31,32,33,34,35
Otras causas de ataxia
Especial interés para el pediatra resulta el conocimiento de que la deficiencia de vitamina E, ya sea adquirida (lactantes prematuros, pacientes con malabsorción intestinal severa), o una forma genética rara (autosómica recesiva) puede causar ataxia, evolucionar de forma progresiva, cuyo curso clínico puede simular una ataxia de Friedreich. El aporte de la vitamina en déficit puede revertir los síntomas o prevenir su progresión.36,37,38
En el curso de un síndrome de malabsorción en un niño, que además se pone atáxico, es importante tener presente la abetalipoproteinemia, enfermedad genética autosómica recesiva del tubo digestivo, que se caracteriza por la ausencia de lipoproteínas de muy baja densidad, mala absorción de las grasas, esteatorrea y puede cursar con retinitis pigmentaria y ataxia.37,38)
Los errores congénitos del metabolismo son una causa común de ataxia.26) Se han descrito más de 150 de ellas en pacientes que además de ataxia presentan un retardo global del neurodesarrollo o su regresión, episodios de encefalopatías, facies tosca, crisis epilépticas y diversos trastornos del movimiento, especialmente convulsiones y regresión del neurodesarrollo; un paciente con ataxia, debe hacer pensar al médico en un error congénito del metabolismo.38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53 Actualmente se dispone de tratamientos, que pueden mejorar la evolución o prevenir la muerte temprana de estos enfermos.27,28,29,30,31,32,33,34,35
Un grupo de enfermedades heredo-degenerativas del sistema nervioso se comportan como una ataxia crónica progresiva, en algunas en el momento solo podemos diagnosticar y tratar sintomáticamente, pero afortunadamente ya en algunas de ellas podemos intervenir y detener el proceso mórbido o alterar positivamente su evolución temporal. Es recomendable tener presente este grupo y hacer un esfuerzo en su diagnóstico y correcto cuidado.54,55,56,57,58
Los tumores primarios del sistema nervioso son la causa más frecuente de tumores sólidos en las edades pediátricas, con una incidencia máxima en la primera década de la vida. Colectivamente representan la segunda causa más frecuente de enfermedades malignas en niños y adolescentes, predominando ligeramente los de fosa posterior (43,2 %), con respecto a los supratentoriales (40,9 %).6,7 Pueden comenzar con una ataxia aparentemente aguda hasta que se hace evidente su curso progresivo: meduloblastomas, astrocitomas, hemangioblastomas cerebelosos. Es importante tener presente esta causa en los niños, donde su diagnóstico puede retrasarse teniendo en cuanta la relativa rareza de estos tumores en ellos, la dificultad de evaluación del niño pequeño y la clínica inespecífica que pueden presentar.
En el niño se pueden expresar todas estas posibilidades etiológicas, unas más frecuentes que otras, pero en todas tenemos la imperiosa necesidad de identificarlas y tratarlas con la mayor prioridad posible, en bien de una evolución favorable o con menos deterioro según la causa.
Consideraciones finales
El cerebelo cumple importantes funciones dentro del sistema nervioso, tiene una expresividad muy típica cuando está enfermo y con el uso adecuado de las nuevas técnicas de estudios por imágenes, genéticas y metabólicas entre otras, permiten al clínico estar en mejores condiciones de su diagnóstico y tratamiento oportuno.

