










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Lowe o síndrome oculocerebrorenal es un trastorno multisitémico, descrito por primera vez en 1952.1 Es una enfermedad extremadamente rara e infrecuente, con una prevalencia estimada en la población general de 1 en 500 000 o 1 a 2000 000 de habitantes, en población caucásica o asiática.2,3
La enfermedad se hereda en forma recesiva ligada al cromosoma X, generalmente solo afecta a los hombres y existen infrecuentes casos descritos en mujeres. Causada por una mutación en el gen que codifica la proteína óculo cerebro renal-Lowe (OCRL1), localizada en Xq26.1, que codifica la enzima fosfatidilinositol bisfosfato 5 fosfatasa. Esta proteína es una inositol fosfato-5-fosfatasa, localizada primordialmente en el aparato de Golgi que afecta diversos procesos celulares incluyendo el transporte transmembrana, remodelación del citoesqueleto de actina, migración celular, polaridad celular y fagocitosis.2,3,4
Clínicamente el síndrome OCRL se presenta con catarata congénita, hipotonía, retraso en el desarrollo neurocognitivo y manifestaciones renales que se desarrollan en los primeros meses o años de vida; asociado a glaucoma congénito. Las manifestaciones renales están dadas por acidosis tubular renal y síndrome de Fanconi y evolución futura a una enfermedad renal crónica. Cognitivamente tienen discapacidad intelectual moderada o severa y el comportamiento obsesivo-compulsivo a menudo está presente, y la epilepsia se establece más tardíamente. También pueden tener manifestaciones clínicas musculo esqueléticas relacionadas con la ERC. El pronóstico es sombrío y la supervivencia es de 30 a 40 años 2,3
El objetivo de este trabajo es exponer un caso clínico típico, con fracaso renal controlado sin método dialítico y que de manera tardía en su adolescencia se diagnostica con síndrome de Lennox-Gastaut.
Presentación del caso
Adolescente de 18 años, primogénito masculino, madre con fórmula gestacional de G4 A2 P2, dos abortos espontáneos sin estudiar y segundo parto de gemelas del sexo femenino, sanas. Embarazo sin riesgo de 40,5 semanas, no hábitos tóxicos, no enfermedades asociadas. Hijo de padres menores de 30 años y no consanguíneos.
Nació de parto eutócico, peso/ 4100 g (macrofeto), talla/50 cm, perímetro cefálico/ 34 cm, Apgar 9/ 9 y opacidad corneal bilateral. Alta hospitalaria al 4to. día sin complicaciones. A los 3 meses de vida, es evaluado en el Hospital Oftalmológico Nacional "Pando Ferrer" por Servicio Nacional de Oftalmología Pediátrica con diagnóstico de catarata congénita, opacidad y edema corneal bilateral. No seguía, ni fijaba objetos y rechazo a luz. No fue tratado quirúrgicamente y se mantuvo conducta expectante. En su evolución clínica, glaucoma severo con compromiso total de la visión.
Desde antes del primer año de vida es atendido en Servicio de Genética y de Nefrología del Hospital Pediátrico “Juan Manuel Márquez”, por hipotonía axial y hiporreflexia osteotendinosa. Tuvo retraso en el desarrollo psicomotor, logró la sedestación y bipedestación después de los 18 meses, con mayor afectación en la esfera motora gruesa y fina.
Con historia de varios ingresos por vómitos, deshidratación y acidosis metabólica, evoluciona con poliuria, trastornos del crecimiento y disminución progresiva de la función renal. En evaluación del servicio de nefrología, por acidosis tubular, trastorno hidroelectrolítico con pérdida de fosfato y calcio por la orina e hipercalciuria, se diagnosticó con síndrome de Fanconi. En evolución a enfermedad renal crónica, etapa II- III por disminución de aclaramiento de creatinina y elevación de la urea plasmática, sin requerir de método dialítico. Presenta raquitismo renal, anemia moderada, aminoaciduria y biopsia renal con glomérulo esclerosis tipo II.
A los dos años se realizó estudio genético en colaboración con el Minsap y centro de referencia en Francia, con estudio molecular de ADN materno y del niño. Se confirma la mutación c2224_2226 del GTA (exón 19), delección de valina en la posición 742 del cromosoma X DEL OCRL 1, que ratifica el síndrome de Lowe.
Desde los tres años en relación con la enfermedad renal crónica avanzada comienza su atención en la consulta externa del Hospital Pediátrico Docente “Centro Habana”, Centro de Referencia Nacional de Nefrología Pediátrica. A los 9 años de edad, presenta disminución significativa del aclaramiento de creatinina compatible con la enfermedad renal crónica terminal y se inicia terapia de reemplazo renal con diálisis peritoneal.
A los 5 meses de recibir tratamiento dialítico, se suspende el tratamiento dialítico por recuperación parcial de la función renal; continuo seguimiento ambulatorio por consulta externa de nefrología, donde se ha mantenido con daño renal correspondiente a una enfermedad renal crónica evolutiva a etapa IV, hasta la actualidad. Con control estricto de la función renal y tratamiento con ingesta libre de agua para compensar las pérdidas urinarias, más soluciones orales de bicarbonato, vitamina D, Ca y eritropoyetina subcutánea; el pronóstico es sombrío por la posibilidad de evolucionar hacia la enfermedad renal crónica terminal que requerirá de método dialítico.
Es visto por primera vez en el Servicio de Neuropediatría y se ingresa a los 10 años, sin crisis previas. Por presentar eventos en horas de 3 a 4 am, con ojos abiertos, "alarido fuerte" y se estira con rigidez de las 4 extremidades en postura de partera de las manos y flexión de miembros inferiores sobre el tronco, duró 2 minutos y repitió en 2 ocasiones en la misma madrugada. Se interpretó como crisis epilépticas generalizadas tónicas durante el sueño. En su evolución clínica tiene crisis de ausencia atípica, atónicas y mioclónicas de miembros superiores, con frecuencia inicialmente mensuales, luego semanales y diarios; de más de 30 crisis al día de varios tipos y predominio de las tónicas durante el sueño. Se realizó electroencefalograma (EEG), sistema internacional 10-20, registro en vigilia y sueño, con trazado característico de punta-onda y poli punta onda variedad lenta, espigas multifocales y atenuación del voltaje. Se confirma por EEG, la presencia de varios tipos de crisis epilépticas y discapacidad intelectual moderada; un síndrome de Lennox-Gastaut (SLG), acompañado de otras manifestaciones neuroconductuales (Fig. 1 [A y B]).
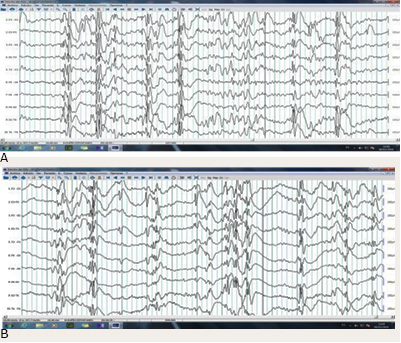
Fig. 1[A y B] - EEG convencional sistema 10-20 sueño espontáneo, etapa N2. Desorganizado a su edad sin grafo elementos y paroxismos de espigas multifocales, puntasondas y poli puntas-onda generalizadas < 2,5 Hz con atenuaciones del voltaje.
Por la severidad de las crisis y difícil control, requirió de poli terapia con 3 drogas. Tratamiento actual con Valproato de sodio a 2000 mg día, en 3 subdosis, Lamotrigina a 200 mg día, 2 subdosis y Clobazan a 30mg día en 2 subdosis. Tiene 8 años de evolución de síndrome epiléptico con control de más de 90 % de las crisis, ya sin crisis tónicas en vigilia y sueño en los últimos 3 años. Las manifestaciones neuroconductuales son: discapacidad intelectual moderada, con coeficiente inteligencia de 45, estereotipias manuales, trastorno de la motricidad fina, obsesión y compulsión por la música, se deprime y frustra con facilidad. Recibe tratamiento con Risperidona. No asiste a ninguna modalidad educacional.
En la actualidad es un adolescente obeso, con Índice masa corporal (IMC) de 32, confinado a un sillón de ruedas. En el examen físico tiene: opacidad corneal, bultalfmos y queloides bilateral con microcefalia, perímetro cefálico/ 50cm con braquicefalia con hipotonía axial, hiporreflexia osteotendinosa y ataxia progresiva (Fig.2).
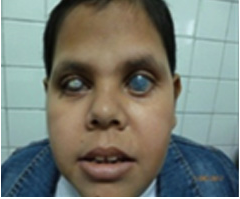
Fig. 2 Bultalfmos bilateral a predominio del ojo izquierdo, con catarata y opacidad corneal bilateral.
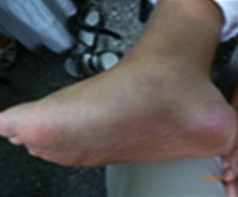
En el tejido celular subcutáneo tiene depósito en partes blandas con aumento de volumen y dolor en zonas de calcáneos y dorsal para vertebral. Con nódulos de tejido celular subcutáneo confirmados por ultrasonido de partes blandas. Tiene una cifo-escoliosis y pie valgus. Relacionado con la hipotonía y miopatía, tiene creatin-phosfokinasa (CPK) (de 450 UI y elevada lactico-deshidrogenesa (LH) (de 1080 UI elevada (Fig. 3).
Ha tenido fracturas patológicas del fémur derecho por hispofosfatemia y fosfaturia asociado por alteraciones del tejido celular subcutáneo antes descrita, ya que no tiene ninguna marcha independiente.
En estudio resonancia magnética nuclear (RMN) de 0.35 Tesla, hay evidencia de imágenes con signos marcados de atrofia cortical generalizada y señales hiperintensa en t2 y flair inespecíficas sugestiva de depósito de aminoácidos e hipoplasia del cuerpo calloso (Fig. 4).
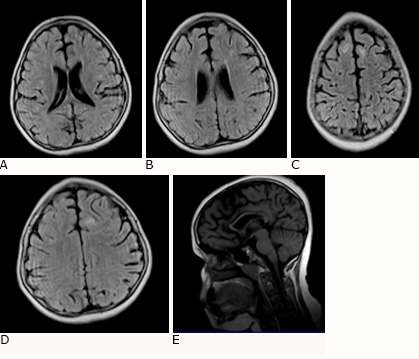
Fig. 4 RMN cráneo T2 flair, corte axial con signos de atrofia cortico-subcortical bilateral, con señales hiperintensas en sustancia blanca (A, B, C, D), corte sagital (E) signos de atrofia cortico-subcortical e hipoplasia del cuerpo callosos.
El paciente ha tenido atención multidisciplinario: servicio nefrología y diálisis. neuropediatría, ortopedia, psiquiatría; apoyo psicopedagógico y tratamiento en relación con las manifestaciones clínicas antes descritas, para lograr una mejor calidad de vida, supervivencia y comenzar su vida adulta.
Discusión
El síndrome de Lowe o síndrome oculocerebrorenal, es una enfermedad multisistémica ligada al cromosoma X, caracterizada por una triada de catarata congénita, discapacidad intelectual y disfunción tubular proximal renal. Los signos clínicos oculares (catarata bilateral y déficit visual) y la hipotonía son las manifestaciones clínicas más significativas desde el nacimiento y antes del primer año de vida. En la primera infancia o edad preescolar, la acidosis tubular con el síndrome de Fanconi y daño tubulointersticial y glomeruloesclererosis renal ya evidencian una progresión a la enfermedad renal crónica; acompañado de retraso en el desarrollo psicomotor y glaucoma severo con bultalfmos y queloides y otras alteraciones cornéales. Es frecuente la discapacidad intelectual con un coeficiente de inteligencia entre 40-54. En la adolescencia y la adultez son ya evidentes las manifestaciones musculo esqueléticas, con deformidad articular, cifosis, escoliosis, fracturas patológicas, depósitos en Tejido celular subcutáneo y ya establecido una enfermedad renal crónica en estadío terminal. El 75 % de los pacientes y en relación con la hipotonía y miopatía logran una marcha independiente, algunos con apoyo entre los 6 a 13 años de edad. Una supervivencia aproximada de 20 a 40 años.1,2,3,4
Las alteraciones de la conducta y el comportamiento se presentan en edad escolar entre los 8 a 13 años. Con irritabilidad, agresividad, estereotipias, trastornos adaptativos y la propia discapacidad intelectual, asociado a conductas repetitivas, síntomas obsesivo-compulsivo, depresión. Con excelente respuesta con neurolépticos, antidepresivos y benzodiacepinas.
La epilepsia no tiene una edad específica de presentación, ocurre en 50 % de los pacientes, no hay evidencia de un tipo particular de crisis epilépticas o síndrome electro clínico definido, generalmente comienza de forma tardía; relacionado con alteraciones estructurales de la sustancia gris y blanca. Las alteraciones neuroradiológicas son inespecíficas demostradas en IRM de cráneo por ventriculomegalia, atrofia cerebral, hipoplasia cerebelosa y trastornos de la migración, desarrollo de la corteza y la mielinización; con depósito de sustancias amorfas o aminoácidos evidentes por lesiones lacunares perivasculares y lesiones de sustancia blanca periventriculares.1,2,3,4
El diagnóstico genético se realiza para determinar la mutación en el gene Xq25-26, ligada al cromosoma X, generalmente afectado en el sexo masculino. Puede haber una sospecha prenatal si se demuestra la catarata y elevación del alfa feto proteína.1,2,3,4
La afectación renal consiste en una disfunción tubular o síndrome de Fanconi que no está presente desde el nacimiento, sino que aparece durante el primer año de vida y se manifiesta como un fallo de medro. Aparece una acidosis tubular proximal, con pérdida de bicarbonato, aminoácidos y fosfatos, poliuria y proteinuria. Como consecuencia, tienen una disminución del aclaramiento de creatinina que los conduce a una enfermedad renal crónica terminal. Los hallazgos en las biopsias renales dependen del estado evolutivo en que se encuentre la lesión. Al inicio, el parénquima renal es normal, luego hay discreta afectación glomerular, que contrasta con la dilatación tubular. El tratamiento es sintomático.1,2,3,4,5
En nuestro caso particular, tiene las características clínicas y evolución clásica del síndrome de Lowe o síndrome oculocerebrorenal. Al nacer con catarata congénita y déficit visual bilateral con hipotonía axial y retraso del desarrollo psicomotor. Desde sus primeros años de vida, las manifestaciones renales de acidosis tubular con síndrome de Fanconi y evolución clínica a una enfermedad renal crónica. En su desarrollo glaucoma severo con los signos descritos en el examen físico. Completa la triada las manifestaciones neuroconductuales, entre ellas la discapacidad intelectual moderada y síntomas obsesivo-compulsivos con depresión. Ya en la adolescencia, manifestaciones musculo esqueléticas hasta una pérdida de la locomoción independiente, con fracturas patológicas, miopatía progresiva y depósitos en el tejido celular subcutáneo, asociado a CPK y LDH elevada.
El síndrome de Lowe, enfermedad renal crónica y síndrome de Lennox-Gastaut, se confirma con el estudio genético realizado en colaboración con centro de referencia en Francia; dada nuestras limitaciones tecnológicas y por el interés especial de nuestro sistema de salud de esclarecer o contribuir al bienestar y desarrollo científico de una entidad genética rara o poco frecuente. Así coincide con otros reportes de casos clínicos2,6,7,8,9 y la identificación de la mutación del OCR-1 en el exón 19, de los más frecuentes entre 200 mutaciones de este gen.7,9
De manera especial su relación con epilepsia y síndrome de Lennox-Gastaut, las series de casos publicados,2,6,7 y las descripciones del síndrome1,3,4 ya mencionado con anterioridad, no establecen una relación definida con edad de inicio, semiología de crisis epiléptica o síndrome epiléptico electro-clínico bien definido.
El síndrome de Lennox-Gastaut es considerado por ILAE (10,11 como una encefalopatía epiléptica de la infancia, entre el 3-10 %, de fisiopatología desconocida y de etiología diversa. Generalmente se inicia entre los 2 a los 8 años (media de 3 a 5 años), con predominio del sexo masculino,12,13,14 aunque en la actualidad hay evidencia hasta los 21 años y edad adulta con menor incidencia.13,14,15,16 Frecuentemente se establece una relación de continuidad con el síndrome de West,12,13,14,15,16,17 que lo precede, su etiología sintomática o probablemente sintomática según clasificación etiológica ILAE (1985-89) (10 y estructural, metabólica, genética, infecciosa, inmunomediada o desconocida vigente desde el 2017;18 aunque en este síndrome de manera particular es más evidente su relación con etiología estructural o genética y metabólica.
Se caracteriza por una triada en la coexistencia de diferentes o varios tipos de crisis, con predominio de las crisis tónicas durante el sueño, ausencias atípicas y drop-attacks (actual crisis atónica) u otras; EEG con punta-onda lenta generalizada, menos de 2,5 Hz y actividad rítmica rápida generalizada durante el sueño (10-20 Hz) o reclutante y retraso del desarrollo psicomotor, acompañado o no de trastornos del comportamiento como discapacidad intelectual y trastorno del espectro autista.12,13,14,15,16,17
Es un síndrome epiléptico de pronóstico reservado o grave, por su pobre respuesta a las drogas antiepilépticas y farmacoresistente. Dado por la severidad, frecuencia y diversidad de crisis epilépticas, su inicio en una etapa importante del neurodesarrollo está precedido por un síndrome de West en alrededor de 30 %, actividad eléctrica característica mantenida y su repercusión en la conducta u comportamiento y esfera cognoscitiva.12,13,14,15,16,17
Todas las características descritas tienen como consecuencia una progresiva disfunción neurológica y alrededor de 5 % fallece y entre 80 a 90 % persiste con crisis en la edad adulta.12,13,14,15,16,17)
Las drogas antiepilépticas de mayor eficacia son el Ácido Valproico, la Lamotrigina, el Clobazam, el Topiramato, el Levetiracetam, la Rufinamida y la Zonisamida. Otras alternativas en caso de refractariedad son: estimulación del nervio vago, dieta cetogénica, estimulación cerebral profunda y uso médico del cannabis,15,17,19,20,21,22 no utilizadas en nuestro contexto clínico.
En nuestro caso es de destacar su relación con una etiología genética y estructural por las alteraciones inespecíficas descritas en la RMN de cráneo, su comienzo tardío y no estar precedido por el síndrome de West. Con una frecuencia al inicio de crisis semanales y luego diarias, con mayor frecuencia de crisis tónica en vigilia y sueño, atónicas, ausencias atípicas y mioclonias. En poli terapia se logró un control de alrededor de 90 % de sus crisis lo que permitió un periodo libre de crisis (3 años) y mejoró su calidad de vida en su transición a la edad adulta. Aunque podría considerase con un pronóstico sombrío por su deterioro neurocognitivo significativo, asociado a otros trastornos del comportamiento, daño multisitémico en asociación a enfermedad renal crónica progresiva todavía sin método dialítico definitivo y otras comorbilidades asociadas. Con enfoque interdisciplinario pudo sobrepasar la edad pediátrica. Su seguimiento, en una institución terciaria como el Hospital Clinicoquirúrgico “Hermanos Ameijeiras”.

