










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
versión On-line ISSN 1561-3062
Rev Cubana Obstet Ginecol vol.42 no.2 Ciudad de la Habana abr.-jun. 2016
REVISIÓN BIBLIOGRÁFICA
De las bases embriológicas a la clínica en el síndrome de Prune Belly
From the embryological basis to the clinical Prune Belly syndrome
Harry Pachajoa
Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras. Universidad Icesi. Cali, Colombia.
RESUMEN
El síndrome de Prune Belly es un trastorno congénito, que obedece según lo reportado actualmente a una base genética. Está caracterizado por la siguiente triada: deficiencia en grados variables de la musculatura abdominal, criptorquidia bilateral y anomalías del tracto urinario. Se identifican dos variantes del síndrome, una mortal y otra compatible con la vida.
Palabras clave: síndrome Prune Belly; obstrucción uretral; ano imperforado, criptorquidia; agenesia abdominal.
ABSTRACT
Prune Belly syndrome is a congenital disorder that is due, as currently reported, to genetic basis. It is characterized by the following triad: deficiency of abdominal muscles in varying degrees, bilateral cryptorchidism and urinary tract anomalies. Two variants of the syndrome are identified, a deadly one and another compatible with life.
Keywords: Prune Belly syndrome; urethral obstruction; imperforate anus, cryptorchidism; abdominal agenesis.
INTRODUCCIÓN
El síndrome de Prune Belly (SPB, MIM: 100100), también conocido como el síndrome de Eagle Barrett o secuencia de Prune Belly, se caracteriza por una triada clásica: ausencia parcial o completa de la pared abdominal, criptorquidia bilateral y anormalidades del tracto urinario.1 Su prevalencia se ha estimado entre 1 en 35,000 y 1 en 50,000 nacimientos. Predomina en el sexo masculino hasta en 97 %, con una proporción de 18:1. 2 El 20 % de todos los pacientes con esta anomalía fallecen en el periodo neonatal como consecuencia de anormalidades del tracto urinario y la hipoplasia pulmonar secundaria a estas3 (cuadro).
Cuadro. Características generales del síndrome del Prune Belly.
| | Hallazgos |
| Características generales | Ausencia parcial o completa de la pared abdominal, criptorquidia bilateral y anormalidades del tracto urinario |
| Sistema urinario | Válvulas uretrales posteriores, Uréteres dilatados, hidronefrosis |
| Sistema Cardiovascular | Ductus arterioso, defectos del septo atrial y tetralogía de Fallot |
| Sistema gastrointestinal | Mal rotación intestinal, atresia anal, estenosis del intestino delgado, gastroquisis y vólvulos |
| Otros | Retardo en el crecimiento, artrogriposis y anormalidades craneofaciales tipo secuencia de Potter |
TEORÍAS Y BASES GENÉTICAS
Se han planteado diferentes teorías acerca de la génesis del SPB; sin embargo, en la actualidad los estudios han apuntado a una base genética.4 Se han discutido genes específicos implicados en la etiología de este síndrome. Tal es el caso de la mutación en el gen del receptor muscarínico colinérgico M3 (CHRM3), localizado en la región 1q43. Está involucrado en el desarrollo de epitelio renal y el músculo de la vejiga y responde a un patrón de herencia autosómico recesivo. El estudio inicial de Weber y otros,5 quienes analizaron a una familia turca consanguínea (cinco hermanos) con el síndrome de Prune Belly, encontró que en ellos existía una mutación homocigótica en el gen CHRM3, y que está relacionada con dicha condición.5 Por otro lado, se ha implicado en el origen del SPB, un factor de transcripción denominado factor nuclear de hepatocitos 1B (HNF 1B), responsable de regular el desarrollo normal del mesodermo y endodermo embrionario.6 El SPB parece ser secundario a la presión ejercida por la expansión de la vejiga o los riñones.7
VARIANTES
Se han descrito dos variantes de este síndrome, en el primer grupo y menos frecuente, la variante letal descrita por Roger y Ostrow en 1973, 7 que corresponde a una uropatia obstructiva secundaria a la presencia de valvas uretrales, que resulta en la muerte perinatal por insuficiencia renal o respiratoria, esta última producto de la hipoplasia pulmonar por oligohidramnios;8 el segundo grupo, corresponde a la forma no obstructiva que se presenta con una anormalidad del vaciamiento vesical, dada por la valsalva insuficiente de los músculos rectos abdominales hipoplasicos, asociado a debilidad del musculo liso de la vejiga.9
DIAGNÓSTICO
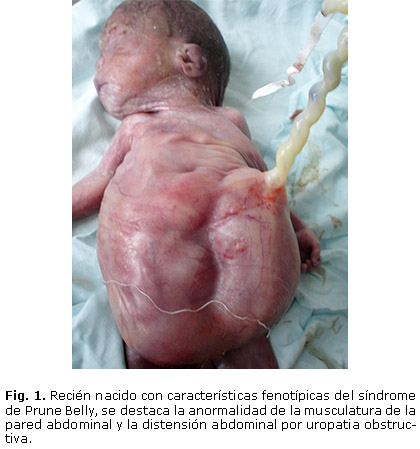
El diagnostico puede ser sospechado prenatalmente en fetos en quienes se presume masas abdominales causadas por una uropatia obstructiva por valvas ureterales o agenesia uretral (figura 1); otras anormalidades del tracto urinario asociadas a este síndrome incluyen uréteres dilatados e hidronefrosis. La displasia renal resulta en oligohidramnios y deformidades relacionadas como la hipoplasia pulmonar que puede llevar a la muerte y deformidades esqueléticas.1, 3 Las alteraciones cardiacas están presentes en menos del 10% de los pacientes; estas y otras anormalidades sistémicas son descritas.
La secuencia de eventos que suceden el SPB son descritos en la figura 2, donde el evento inicial es la obstrucción temprana de las vías urinarias que va desencadenando los eventos propios de la triada clásica. El defecto de la musculatura de la pared abdominal en el SPB parece ser secundario a la presión ejercida por la expansión de la vejiga o los riñones.7
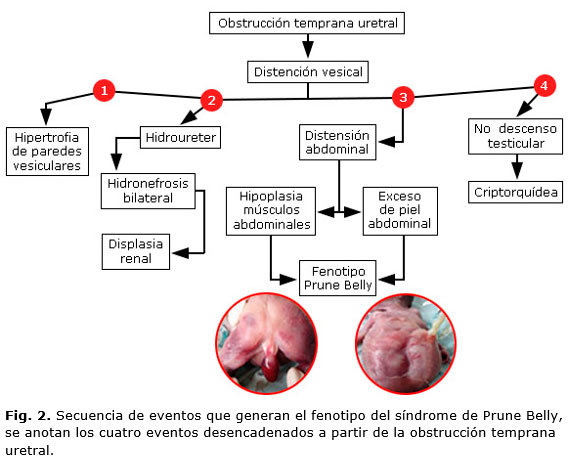
Los hallazgos fenotípicos del paciente con SPB de la figura 2, son ejemplos de la variante letal del SPB, que forma parte del grupo de enfermedades que producen la uropatia obstructiva. Esta causa graves alteraciones fetales y muertes perinatales, por lo que se hace necesario más estudios encaminados a entender la fisiopatología de esta condición para iniciar un manejo integral rápido in útero. Además de un diagnóstico temprano, donde se incluya un asesoramiento genético.
Conflicto de interéses
Las autoras no delcaran conflictos de interéses.
REFERENCIAS BIBLIOGRÁFICAS
1. Poucell-Hatton S, Huang M, Bannykh S, Benirshke K, and Masliah E: Fetal obstructive uropathy: Patterns of renal pathology. Pediatric and Developmental Pathology. 2000;3:223-31.
2. Greskovich FJ, Nyberg LM. The Prune Belly syndrome: A review of its etiology, defects, treatment and prognosis. J Urol. 1988;140:707-12.
3. Stephens, FD, Gupta, D. Pathogenesis of the prune belly syndrome. J Urol. 1994;152:2328-31.
4. OMIM Entry - # 100100 - Abdominal muscles, absence of, with urinary tract abnormality and cryptorchidism [Internet]. [Citado 2 de octubre de 2015]. Disponible en: http://www.omim.org/entry/100100.
5. Weber S, Thiele H, Mir S, Toliat MR, Sozeri B, Reutter H, Woolf AS. Muscarinic acetylcholine receptor M3 mutation causes urinary bladder disease and a prune-belly-like syndrome. The American Journal of Human Genetics. 2011;89(5):668-74.
6. Granberg CF, Harrison SM, Dajusta D, Zhang S, Hajarnis S, Igarashi P, Baker LA. Genetic basis of prune belly syndrome: screening for HNF1β gene. The Journal of Urology. 2012;187(1):272-8.
7. Laborie LB, Mackay DJ, Temple IK, Molven A, Sovik O, Njolstad. DNA hypomethylation, transient neonatal diabetes, and prune belly sequence in one of two identical twins. Eur J Pediatr. 2010;169(2):207-13.
8. Haeri S, Devers PL, Kaiser-Rogers KA, Moylan Jr VJ, Torchia BS, Horton AL, Aylsworth AS. Deletion of hepatocyte nuclear factor-1-beta in an infant with prune belly syndrome. American Journal of Perinatology. 2010;27(7):559-63.
9. Rogers LW, Ostrow PT. The Prune Belly syndrome. Report of 20 cases and description of a lethal variant. J Pediatr. 1973;83:786-93.
Recibido: 29 noviembre 2015.
Aprobado: 3 febrero 2016.
Harry Pachajoa. Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras. Universidad Icesi. Cali, Colombia.

