










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
versión On-line ISSN 1561-3062
Rev Cubana Obstet Ginecol vol.44 no.1 Ciudad de la Habana ene.-mar. 2018
REVISIÓN BIBLIOGRÁFICA
Asociación de defectos cardíacos con síndromes genéticos y su utilidad en el diagnóstico prenatal
Association of heart defects with genetic syndromes: utility in prenatal diagnosis
Yamilet Corona Carnero,I Al. Raúl Ernesto Pérez Corona,II Al. Leydis Yamilet Fiallo DelgadoIII
I Departamento de Genética. Municipio Habana del Este. La Habana, Cuba.
II Facultad de Ciencias Médicas Enrique Cabrera. La Habana, Cuba.
III Facultad de Ciencias Médicas Victoria de Girón. La Habana, Cuba.
RESUMEN
Se realiza una revisión bibliográfica de los síndromes genéticos que se asocian a defectos cardíacos por ser las cardiopatías congénitas causa frecuente de mortalidad neonatal y primer año de vida. Si las mismas se asocian a síndromes genéticos, la supervivencia se reduce a más del 70 %. En 85 % de los defectos cardíacos existen evidencias de la asociación a síndromes genéticos y un 15 % de ellos puede relacionarse con una herencia monogénica o una anomalía cromosómica. Estos síndromes presentan otras malformaciones visibles por el examen ecográfico, que ayudan a realizar prenatalmente el diagnóstico de estos. Síndromes tales como Alagille, Down, Holt-Oram, DiGeorge, Costello; se asocian a defectos cardíacos como las alteraciones en la septación atrioventricular o de los tractos de salida. Pensando en esta asociación podemos realizar un diagnóstico certero y un asesoramiento adecuado de la pareja que se enfrenta a tal situación.
Palabras clave: diagnóstico prenatal; síndromes genéticos; defectos cardíacos.
ABSTRACT
Congenital heart diseases are a common cause of death in the neonatal stages and first year of life, but in association with genetics syndromes the possibility of survivor decreases in 70 percent. A review on the subject is carried out in order to broaden the interpretation of cardiac defects in the ultrasound diagnosis and the association with other malformations and genetic syndromes. In 85 % of cardiac defects there is evidence of the association to genetic syndromes and 15 % of them can be related to a monogenic inheritance or a chromosomal anomaly. These syndromes present other malformations visible in the ultrasound examination, which help prenatal diagnosis. Alagille, Down, Holt-Oram, DiGeorge, Costello Syndromes are associated with heart defects such as alterations in atrioventricular septation or outflow tracts. Reviewing this association we can make an accurate diagnosis and adequate advice for the couple who are facing such a situation.
Keywords: prenatal diagnosis; genetic syndromes; heart defects.
INTRODUCCIÓN
Dentro del diagnóstico prenatal, los defectos cardíacos se encuentran entre las malformaciones más frecuentes, con una incidencia de casi 1 de cada 100 nacidos vivos. Para los especialistas que se dedican a la detección intrauterina de defectos fetales, suele ser el sistema cardiovascular el de más difícil diagnóstico prenatal, por las características ya conocidas de éste en la vida intrauterina.1,2
Las cardiopatías congénitas son causa frecuente de defunción en las etapas neonatales y primer año de vida. Con los adelantos de la ecografía, cada vez es más probable el diagnóstico certero de estos defectos congénitos que permiten un actuar oportuno para la interrupción del embarazo o la planificación de las posibles soluciones al defecto en el recién nacido.3
Muchas veces, en la premura del diagnóstico y la rápida solución al problema, se queda rezagado el estudio y análisis completo de lo que más allá de este acontecimiento puede estar sucediendo en el feto, como son las asociaciones de las alteraciones cardíacas a otros defectos y a su vez la presencia de síndromes genéticos, que de no ser diagnosticados, estudiados consecuentemente e informados a las parejas afectadas, repercuten en la ayuda que le podemos brindar para futuras decisiones y posible planificación de nuevos descendientes en ellas.4
Se ha demostrado que la interferencia en las funciones de muchos genes puede traer como consecuencia una amplia gama de defectos cardíacos y vasculares. Pero la base de los defectos cardíacos también puede relacionarse con la alteración de una amplia variedad de moléculas, que tienen responsabilidad en la cascada de la formación normal de una determinada parte del corazón fetal.5
Si partimos del hecho de que el organismo humano funciona como un todo desde que comienzan los primeros estadíos de su formación, podemos pensar que una alteración en uno de sus genes o desde el punto de vista molecular puede llevar a defectos de varios órganos o sistemas de órganos que se pueden relacionar como un síndrome genético en la mayor parte de los casos. En varias investigaciones ha quedado claro que el mismo gen puede actuar en períodos diferentes de todo el proceso de formación del feto y en órganos distintos, así mismo ocurre con las bases moleculares que intervienen eficazmente en la diferenciación de órganos.5
El objetivo de la revisión es ampliar la interpretación de los defectos cardíacos en el diagnóstico ecográfico y su asociación a otras malformaciones y síndromes genéticos. Al estar ante uno de estos casos, esperamos que no sean diagnosticados la mayor parte de las veces como defectos aislados; sino que pensemos al encontrarlos, en buscar asociaciones que podrían pasar desapercibidas por el ecografista. De esta forma se podrá enriquecer el diagnóstico y aumentar las posibilidades de asesoramiento a la pareja. También se realizará una breve revisión de los principales defectos cardíacos diagnosticables prenatalmente y su asociación a síndrome genéticos.
ASOCIACIÓN DE DEFECTOS CARDIACOS CON SÍNDROMES GENÉTICOS
Cómo determinar en el estudio ecográfico la posición del corazón fetal
Cuando comenzamos a realizar el examen ecográfico fetal, debemos primeramente identificar el lado derecho e izquierdo del feto mediante la determinación de la posición de la columna vertebral y la cabeza. En un corte transversal, determinamos el situs visceral fetal, lo que demuestra la posición del estómago, vasos hepáticos, aorta abdominal y vena cava inferior. Con este previo análisis, estamos en condiciones de hallar la posición del corazón fetal, para conocer si estamos en presencia de un "situs solitus" o realizar el diagnóstico de un "situs inversus".6 A continuación, realizamos algunas consideraciones acerca de la rotación cardíaca y sus posibles alteraciones.
Síndromes cardioesplénicos: asplenia-poliesplenia
Son alteraciones en la lateralización de la asimetría corporal normal, que abarcan defectos cardíacos graves y anomalías en las vísceras intratorácicas e intraabdominales. Los síndromes de heterotaxia se caracterizan por la detección bilateral de órganos que en condiciones normales se encuentran del lado izquierdo (poliesplenia) o por el hallazgo bilateral de órganos que en condiciones normales se encuentran del lado derecho (asplenia) y es probable que sean dos manifestaciones distintas de una alteración en la lateralización de la asimetría corporal normal. Ambos síndromes se asocian a anomalías cardíacas graves, aunque los que presentan asplenia podrían ser más severos.6
Recordemos que, hasta el momento de la gastrulación, el embrión es bilateralmente simétrico y al comenzar esta, se ponen una serie de mecanismos en marcha que provocan la incurvación del tubo cardíaco hacia la derecha; seguida de una incurvación asimétrica del tubo digestivo, lo que hace posible la asimetría también de hígado, bazo y la lobulación de los pulmones.7
El mecanismo que explica la aparición de asimetría en el embrión consta de varios factores. Entre ellos se encuentran mecanismos moleculares como la expresión de las moléculas de señalización nodal, cuya función es la ruptura de simetría en su lado izquierdo, y por otro lado; lefty-1, encargada de evitar la difusión de las moléculas que determinan la izquierda en el lado derecho del embrión.8
Otro mecanismo que puede provocar el síndrome cardioesplénico es la falta de activación del gen Pitx2, factor de transcripción involucrado en la rotación del intestino y el estómago, la posición del hígado y el bazo, así como la lobulación asimétrica de los pulmones.9
La polaridad anteroposterior, mostrada por la línea primitiva con la participación de la polaridad celular plana, mecanismo que involucra las proteínas Dishevelled y Prickle, aún en investigación, podría también explicar la aparición de los trastornos en la asimetría corporal.10,11
Las anomalías que se pueden asociar a estos son las siguientes:
- Vértice cardíaco a la derecha,
- DATVP,
- VCSI persistente,
- Defectos de las almohadillas endocárdicas,
- TGV,
- Ventrículo único,
- Estenosis pulmonar,
- Continuidad de la vena ácigos en la VCI.
El situs in versus puede ser total, como ocurre en el Síndrome de Kartagener (anomalías del seno en situs inverso) o parcial donde solo encontraremos la presencia de Dextrocardia.5
Dextrorotación del corazón
Es frecuente ver asociada la dextrorotación cardíaca a la hipoplasia pulmonar. Los pulmones fetales necesitan para su desarrollo la presencia del líquido en su interior, que la mayor parte de este es producido por las células epiteliales del pulmón fetal. Existe una clara relación entre la presencia de este líquido pulmonar y los movimientos respiratorios, en la que participan también la laringe con una función tipo valvular.
El feto comienza a realizar claros movimientos respiratorios desde las 10 semanas de gestación y son el principal factor estimulante del crecimiento de los pulmones fetales, pues estimulan en este la producción de factor de crecimiento derivado de las plaquetas, factor de crecimiento similar a la insulina y factor de transcripción tiroideo, que, a su vez, intervienen en la proliferación celular y reducen la apoptosis en las porciones periféricas de los pulmones fetales.12,13
Al existir una hipoplasia pulmonar, se encuentra generalmente la Dextrorotación cardíaca, como puede ser observada en el Síndrome de la cimitarra, donde además de esta existe una hipoplasia del pulmón derecho. Este síndrome, por demás infrecuente, se acompaña de drenaje anómalo de venas pulmonares y puede ser diagnosticado a partir de las 20 semanas de gestación. Puede encontrarse el pulmón derecho pequeño, dextroposición cardíaca, arteria pulmonar distendida en relación con la aorta, una rama derecha de la arteria pulmonar con diámetro disminuido y en ocasiones, por la presencia de una vena cava superior izquierda persistente drenando en el seno coronario. Su etiología y herencia aún son desconocidas.14-16
Existen otras alteraciones fetales que pueden provocar desplazamiento del corazón fetal hacia la derecha como las hernias diafragmáticas, masas intratorácicas, derrame pleural y enfermedad adenomatoidea quística pulmonar.17
Defectos en la septación cardíaca
Defectos del tabique interauricular: Las variantes anatómicas más frecuentes que se reconocen como causantes de este defecto son las debidas a una reabsorción excesiva del tejido que rodea el foramen secundum o un crecimiento hipoplásico del septum secundum, aunque también puede ser provocada por la falta de unión entre el borde libre del septum primun y los cojinetes endocárdicos. Existen muchas variantes de este defecto; que puede ir desde una aurícula única, defecto este grave y generalmente asociado a otras malformaciones, hasta la presencia de canal auriculoventricular.18
Los genes que participan en la formación del tabique interauricular son numerosos (Nkx2-5, GATA4 y Tbx5). Los defectos septales auriculares se asociancon frecuenciacon alteraciones del cromosoma 21. La relación entre la Trisomía 21 y los defectos septales entre aurículas y ventrículos es común, además de una relación estrecha con la presencia de canal auriculoventricular.8 Es por esta razón, que al diagnosticar un defecto septal, el ecografista debe buscar además otros hallazgos que puedan ayudar al diagnóstico del síndrome de Down: pliegue nucal aumentado (Fig. 1), hipoplasia del hueso nasal, ecogenicidad intestinal aumentada, signos de atresia duodenal, clinodactilia, orejas pequeñas, entre otros.19-21
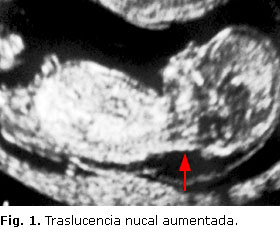
El síndrome de Holt-Oram, trastorno autosómico dominante, se asocia frecuentemente a defectos septales auriculares y ventriculares (Fig. 2). En este existe una mutación del Tbx-5 del gen T-box y se asocia a anomalías de los miembros; en este caso, hipoplasia del radio, pulgares ausentes, sindactilia, focomelia, anomalías claviculares y humerales.22-24
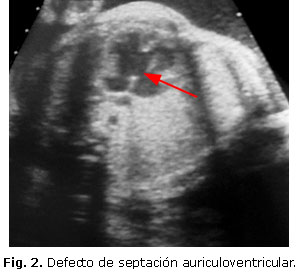
Existen otros síndromes asociados a defectos septales como son el síndrome de Noonan y el Turner, donde generalmente se realizan diagnósticos precoces antes de poder detectar defectos de septación cardíaca, pues es frecuente que se relacionen con aumento de la traslucencia nucal en el primer trimestre, higromas quísticos pequeños en la región lateral del cuello encontrados en el II trimestre de la gestación.25-27
Los defectos de septación ventricular representan el defecto cardíaco más frecuente en los niños, casi 70 % afecta a la porción membranosa del tabique interventricular. Muchas de las mutaciones causantes de defecto en el tabique interauricular, pueden ser causa además de alteraciones en la formación del tabique interventricular.28,29
El síndrome de Di George, asociado a una deleción del cromosoma 22, se asocia a este defecto y puede sospecharse por el ecografista, al realizar la vista extendida de los tres vasos (Fig. 3); donde confirmará la hipoplasia tímica que acompaña a este síndrome y se impondría además la búsqueda de paladar hendido, microcefalia, micrognatia, entre otras anomalías. Es importante destacar, que como veremos más adelante, este síndrome se puede asociar a múltiples defectos cardíacos, la mayoría de ellos se corresponden con cardiopatías congénitas complejas.30-32
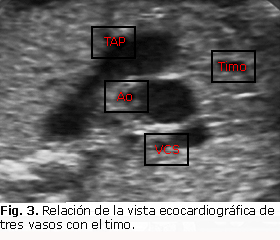
Otros de los síndromes asociados a este defecto cardíaco es el denominado CHILD; enfermedad rara, monogénica, que se trasmite con un rasgo dominante ligado al cromosoma X y causado por mutación en el gen NSDHL. Se sospecha cuando además del defecto cardíaco encontrado existen alteraciones en las extremidades (alteraciones en los dedos, ausencia de una extremidad, hipoplasia de mandíbula, clavícula, escápula) y también asociado a agenesia renal unilateral.33
Malformaciones del tracto de salida
Puede sufrir múltiples malformaciones las que representan entre 20 y 30 % de todas ellas. En la formación de la región troncoconal existe una destacada contribución de las células de la cresta neural. Algunos de los trastornos del tracto de salida se asocian a translocaciones o deleciones del cromosoma 22, al cual hicimos referencia en el párrafo anterior. Desde el punto de vista molecular, se ha investigado que muchas mutaciones en alguno de los componentes de la cascada del factor de crecimiento nervioso, como endotelina-1, Hand-2 o neuropilina-1. Este último receptor para el factor de crecimiento endotelial, además, pueden provocar grados variables de alteraciones en el tracto de salida.
El tronco arterioso persistente se produce por la ausencia de división del tracto de salida mediante las crestas troncoconales y como estas también son encargadas de contribuir a la formación de la porción membranosa del tabique interventricular. Este defecto se acompaña de defectos en él; por lo que se puede encontrar en cualquiera de los síndromes genéticos mencionados en los defectos de septación.34
Otro de los defectos cardíacos diagnosticable prenatalmente es la transposición de grandes vasos, donde se observa la aorta que sale del ventrículo derecho y la arteria pulmonar, a su vez, del ventrículo izquierdo. Este defecto es explicable porque en ocasiones, las crestas trococonales no toman su forma espiral para dividir el tracto de salida en dos conductos, que luego se convierten en dos arcos circulatorios completamente independientes. Es de fácil diagnóstico prenatal porque en la vista de eje largo del ventrículo izquierdo, observaremos la típica imagen en doble cañón de escopeta (Fig. 4) y en la vista de tres vasos podremos apreciar la arteria aorta más anterior que la arteria pulmonar. Se ha visto frecuentemente asociada al Síndrome velo-cardio-facial o Di George.35,5
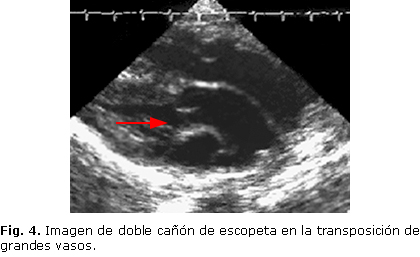
Con una incidencia de 1: 7 000-9 000 nacidos vivos, la Trisomía 13 o síndrome de Patau, puede estar relacionado con este defecto cardíaco; donde en la etapa fetal, encontraremos además la presencia de labio leporino, fisura palatina, microcefalia, micrognatia, defectos en la pared abdominal, riñones poliquísticos, polidactilia entre otras alteraciones.36
La estenosis aórtica y pulmonar ocurren al producirse una tabicación asimétrica del tracto de salida por parte de las crestas trococonales, haciendo que se estreche de forma anómala la arteria aorta o la pulmonar, pudiendo llegar a ser tan severa en algunos casos, que la luz del vaso está básicamente obliterada, denominándose atresia aórtica o pulmonar según sea el caso. En el síndrome de Waardenburg Tipo I, donde existe una mutación en PAX-3, se producen una serie de combinaciones que pueden afectar la cresta neural cardíaca en migración por lo que puede asociarse a defectos en el tracto de salida cardiovascular. Por lo que al existir una estenosis aortica o pulmonar; debemos buscar además la presencia de hipertelorismo, paladar hendido.37,38
En la estenosis pulmonar sospecharemos el diagnóstico ecocardiográfico prenatal cuando en la vista de cuatro cámaras encontramos predomino de cavidades derechas, hipoplasia de la arteria pulmonar y dilatación postestenótica.17
Entre los síndromes genéticos que pueden relacionarse con estas alteraciones, se encuentra el ya mencionado síndrome de Noonan, frecuentemente acompañado de estenosis pulmonar. Los síndromes de William y Marfán, se asocian a estenosis valvular aórtica. En este último, síndrome con patrón de herencia autosómico dominante; donde se cree que puede existir mutaciones en el gen de la fibrilina FBN1, pueden hallarse prenatalmente: catarata, paladar hendido, escoliosis y hemivértebras.39,40
Podemos referirnos en este momento, en que hicimos referencia a la estenosis pulmonar, a una malformación cardíaca que incluye este defecto: tetralogía de Fallot. (Fig. 5) Esta malformación representa del 5 al 10 % de las cardiopatías congénitas y se relaciona con una variedad de alteraciones cardíacas, extracardíacas y cromosómicas, entre 12 y 50 % de los casos. Esta malformación aparece en un 50 % de los casos con deleción 10q, en 40 % de las trisomías 13,25 % de las trisomías 18 y en un 5 a 10 % de las correspondientes al cromosoma 21.17
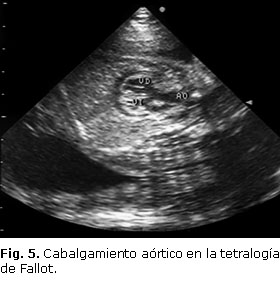
En esta cardiopatía, además de estar presente la estenosis pulmonar infundibular, encontraremos: defecto del tabique interventricular membranoso (subaórtico), cabalgamiento aórtico e hipertrofia de ventrículo derecho. El defecto básico en esta afección es una fusión asimétrica de las crestas troncoconales y una mala alineación de las válvulas aórtica y pulmonar.5,41
Para realizar el diagnóstico prenatal de esta cardiopatía, el ecografista deberá realizar las diferentes vistas ecocardiográficas que demostrarán los defectos ya mencionados. En la vista de cuatro cámaras observará el aumento de cavidades derechas. Es de gran utilidad realizar la vista ecocardiográfica de los tres vasos donde observará la presencia de una arteria aorta más grande que la pulmonar, en ocasiones se acompaña de atresia de la válvula pulmonar, en este caso será difícil visualizar el TAP. El cabalgamiento aórtico, característico de esta malformación, será comprobado al realizar la vista correspondiente al eje largo del ventrículo izquierdo. Al realizar la vista del eje corto de la aorta, comprobaremos el desplazamiento infundibular que acompaña a esta cardiopatía.3
El síndrome de Alagille, causado por una mutación de Jagged-1, un ligando del receptor Notch, se encuentra comúnmente relacionado con la tetralogía de Fallot. Este síndrome se caracteriza además por la presencia de atresia biliar, sospechada siempre ante la ausencia de la vesícula durante varios estudios ecográficos.17
Otra de las asociaciones descritas en relación con esta malformación es el Síndrome de Costello. Las referencias bibliográficas actuales lo describen dentro de los síndromes neuro-cardio-facio-cutáneos, caracterizados por presentar rasgosdismórficos muy semejantes, cariotipo normal y base etiopatogénica e incluso molecular común, determinada por hiperactivación de la vía de transducción Ras-MAPK, resultado de una mutación en el protooncogen HARS. Este síndrome se asocia a defectos en los tabiques interauriculares e interventriculares, estenosis pulmonar y tetralogía de Fallot, entre otras.42
Como mencionamos en párrafos anteriores, esta cardiopatía está asociado frecuentemente a otros síndromes ya mencionados anteriormente como las trisomías 13, 18 y 21 además de asociarse con el síndrome de Holt-Oram; por lo que, al sospecharla, el ecografista debe además realizar un examen exhaustivo para hallar otras de las alteraciones prenatales que caracterizan los síndromes antes expuestos.43
En un examen ecográfico prenatal del corazón fetal, también podemos diagnosticar defectos en la formación del arco aórtico; para lo cual resulta de gran utilidad realizar la vista ecocardiográfica de los tres vasos. Entre las malformaciones más frecuentes se encuentra la coartación de la aorta, con sus dos variantes preductal y posductal. La embriogénesis de la cuál no se encuentra muy bien descrita, pero si su asociación a síndromes genéticos como la Trisomía 21, el Síndrome de DiGeorge y el Turner.44
Al realizar la vista ecocardiográfica de cuatro cámaras encontraremos un aumento de las cavidades derechas y en los tres vasos observaremos una arteria aorta de pequeño calibre, si realizamos una búsqueda del arco aórtico podremos observar un istmo hipoplásico que encuentra presente en el 80 por ciento de las coartaciones. Al realizar la vista ecocardiográfica del eje largo de la aorta y utilizar estudio Doppler, se podrá identificar el segmento hipoplásico.45
La presencia de tumores intracardiacos sonmasas casi siempre múltiples que pueden comprometer el endocardio, miocardio o epicardio. Los que más frecuentemente se reportan son del tipo Rabdomiomas, aunque también existe incidencia reportada de fibromas y teratomas. Constituyen una alteración de fácil diagnóstico al realizar la vista ecocardiográfica de cuatro cámaras. También puede encontrarse asociada a enfermedad congénita, en este caso, esclerosis tuberosa. El patrón de herencia de esta afección es autosómico dominante, aunque dos terceras partes de los casos son mutaciones nuevas en familias no comprometidas. En el diagnóstico de esta afección suele ser el defecto cardíaco, el primer signo que hace sospecharla. Se informa que entre 40 y 68% de los fetos con Rabdomiomas cardíacos pueden presentar esta enfermedad.46
CONCLUSIONES
El estudio ecográfico, cada vez con más avances en el diagnóstico prenatal, permite la descripción segura de la anatomía fetal normal. Además, avizora una imagen más certera de la posible evolución del feto, lo que descarta la presencia de defectos congénitos. Si sumamos a esto la idea de establecer asociaciones a través del diagnóstico prenatal con los diferentes síndromes genéticos; el margen de error y la posibilidad de que nos sorprenda un feto con malformaciones, será cada vez menor. Del total de defectos cardiacos, 85 % involucran factores genéticos y en un 15 por ciento de ellos existe una herencia monogénica o una anomalía cromosómica. Conocer de las asociaciones de los defectos cardíacos con síndromes genéticos nos amplía las posibilidades diagnósticas y además ayuda al asesoramiento de la pareja aumentando la calidad y la integralidad en la atención prenatal.
Tener una visión integral del feto nos ayuda a diagnosticar con mayor frecuencia la presencia de síndromes genéticos. Aún más, nos asegura la calidad de vida del futuro bebé; lo que constituye el principal regocijo para el especialista en su quehacer diario.
REFERENCIAS BIBLIOGRÁFICAS
1. Anuario Estadístico de Salud 2010. Dirección Nacional de Registros Médicos y Estadísticas de Salud. La Habana: Ministerio de Salud Pública; 2011.
2. Rugieri VL, Arberas CL. Síndromes genéticos reconocibles en el período prenatal. Medicina (Buenos Aires) Revista en Internet. 2009[citado 14 Ene 2013];31(Suppl 2):aprox. 40 p. Disponible en: http://scielo.isciii.es.php?script=sciarttext&pid=S1137-66272008000400004
3. García Guevara C, Arencibia Faife J, Savío Benavides A. Valor de la vista ecográfica de los tres vasos en el pesquisaje de cardiopatías congénitas. Rev Cubana Genet Comunit. 2010[citado 15 Ene 2013];4(1):5-9.
4. Alonso LF. Características etiopatogénicas de los recién nacidos con malformaciones múltiples. Rev Cubana Pediatr. 1998[citado 14 Ene 2013];70(2):73-8.
5. Carlson BM. Embriología humana y biología del desarrollo. España: Elsevier Saunders; 2014[citado 14 Ene 2013];58-73.
6. Berg C, Geipel A, Khol T. Fetal echocardiographic evaluation of atrial morphology and the prediction of laterality in case of heterotaxysyndroms. Ultrasound Obstet Gynecol. 2005[citado 15 Ene 2013];26:538-45.
7. Chapell JC, Bautch VI: Vascular development: genetics mechanism and links to vascular disease. Curr Top Dev Biol. 2010[citado 14 Ene 2013];90:43-72.
8. Garg V: Insights into the genetic basis of congenital heart disease. Cell Mol Life Sci. 2006[citado 14 Ene 2013];63:1141-48.
9. Clark KL, Yutzey KE, Benson DW. Transcription factors and congenital heart defects, Annu Rev Physiol. 2006[citado 16 Ene 2013];68:97-121.
10. Casey B, Hackett BP. Left axis malformation in man and mouse.Dev CurrOpin Genet. 2000[citado 16 Ene 2013];10:257-61.
11. Levin M.Left-right asymmetry in embryonic development: a comprehensive review. Mech Dev. 2005[citado 15 Ene 2013];122:3-25.
12. Wallingford JB. Planar cell polarity signally, cilia and polarized ciliary beating. CurrOpin Cell Biol. 2010[citado 14 Ene 2013];22:597-604.
13. Cook Ac, Yates RW, Anderson RH. Normal and abnormal fetal cardiac anatomy, PrenatDiagn. 2004[Citado 15 Ene 2013];24:1032-48.
14. Inanlou MR, Baguma-Nibasheka M, Kablar B. The role on the fetal breathing-like movements in lung organogenesis, HistolHystopathol. 2005[Citado 14 Ene 2013];20:1261-6.
15. Abdullah MM, Lacro RV, Smallhorn J. Fetal cardiac dextroposition in the absence of an intrathotacic mass; sign of significant right luhghypoplaisa. J Ultrasound Med. 2000[Citado 14 Ene 2013];19:669-76.
16. Grisaru D, Achiron R, Lipitz S.Antenatal sonographic findings associated whit scimitar syndrome. Ultrasound Obster Gynecol. 1996[Citado 14 Ene 2013];8:131-3.
17. Michailidis GD, Simpson JM, Tulloh RM. Retrospective prenatal diagnosis of scimitar syndrome aided by three-dimensional power Doppler imaging, Ultrasound Obstet Gynecol. 2001[Citado 14 Ene 2013];17:449-52.
18. Oliva Rodríguez JE. Ultrasonografía diagnóstica fetal, obstétrica y ginecológica. Ecimed: La Habana. 2010;6:119-21.
19. Rudolfph AM. Congenital cardiovascular malformation and the fetal circulation. Arch Dis Child Fetal Neonatal. 2010[citado 14 Ene 2013];95:132-6.
20. Bahado-Singh RO, Goldstein I, Uerpairojkit B. Normal nucal thickness in the midtrimester indicates reduce risk of Down syndrome in pregnancies whit abnormal triple-screen results. Am J Obstet Gynecol. 1995[citado 15 Ene 2013];173:1106-10.
21. Benacerraf BR, Baras VA, Laboda LA. A sonographic sign for the detection in the second trimester of the fetus whit Down syndrome. Am J Obstet Gynecol. 1985[citado 15 Ene 2013];151:1078-9.
22. Bromley B, Lieberman E, Shipp TD. The genetic sonogram, a method for risk assessment for Down syndrome in the mid trimester. Ultrasound Med. 2002[citado 15 Ene 2013];21:1087-96.
23. Basson CT, Cowley GS, Solomon SD. The clinical and genetic spectrum of the Holt-Oram síndrome(heart-hand síndrome). N Engl J Med. 1994[citado 16 Ene 2013];330:885-91.
24. Holt M, Oram S. Familial heart disease whit skeletal malformations. Br Heart J. 1960[citado 16 Ene 2013];22:236-42.
25. Tongsong T, Chanprapaph P. Prenatal dyagnosis of Holt-Oram syndrome. J Clin Ultrasound. 2000[citado 16 Ene 2013];28:98-100.
26. Donnenfel AE, Nazir MA, Sindoni F. Prenatal sonographic documentation of cystic higroma regression in Noonan síndrome. Am J Med Genet. 2001[citado 14 Ene 2013];39:461-5.
27. Menashe M, Arbel R, Raveh D. Poor prenatal detection rate of cardiac anomalies in Noonan syndrome. Ultrasound Obstet Gynecol. 2002[citado 14 Ene 2013];19:51-5.
28. Bernstein HS, Filly RA, Goldberg JD. Prognosis of fetuse whit a cystic hygroma. PrenatDiagn. 1991[citado 14 Ene 2013];11:349-55.
29. Allan LD, R Santos, Pexeider P (1997): Anathomical and ecocardiographic correlations of normal cardiac morphology in the late first trimester fetus. Heart[citado 14 Ene 2013];77:68-72.
30. De León Ojeda NE, Pérez Mateo MT, Ramiro Novoa JC. Evaluación de los criterios de indicación y positividad de la ecocardiografía fetal en gestantes de alto riesgo. Rev Cubana Genet Comunit. 2007[citado 14 Ene 2013];1(1):25-9.
31. Chaoui R, Kalache KD, Heling KS. Absent or hypoplastic thymus on ultrasound: a maker for deletion 22q11 in fetal cardiac defects. Ultrasound Obstet Gynecol. 2002[citado 14 Ene 2013];20:546-52.
32. Moore JW, Binder GA, Berry R. Prenatal diagnosis of aneuploidy and deletion 22q11 in fetus whit ultrasound detection of cardiac defects. Am J Obstet Gynecol. 2004[citado 14 Ene 2013];191:2068-73.
33. Sleurs E, De Catte L, Benatar A. Prenatal diagnosis of absent pulmonary valve síndrome in association whit 22q11 deletion. J Ultrasound Med. 2004[citado 13 Ene 2013];23:417-22.
34. Morilla Guzmán A, de León Ojeda NE, García del Collado AE, Petizco Hernández A. Presentación de un caso de hemidisplasia congénita con ictiosis eritrodérmica. Rev Cubana Pediatr. 2008[citado 13 Ene 2013];80(3).
35. Rentschler S, Jain R, Epstein JA. Tissue-tissue interactions during morphogenesis of the outflow tract.PediatrCardiol. 2010[citado 13 Ene 2013];31:408-13.
36. Chen CP, Chern SR, Chang TY. Prenatal diagnosis of mosaic ring chromosome 22 associated whit cardiovascular abnormalities and intrauterine growth restriction. Prenat Diagn. 2003[citado 14 Ene 2013];23:40-3.
37. Benacerraf BR, Miller WA, Frigoletto FD. Sonographic detection of fetuses whit trisomy 13 and 18: accuracy and limitations. Am J Obstet Gynecol. 1988[citado 13 Ene 2013];159:404-9.
38. Meulemans D, Bronner-Fraser M: Gene-regulatory interactions in neural crest evolution and development. Dev Cell. 2005[citado 14 Ene 2013];7:291-9.
39. Kulesa PM, Gammill LS. Neural crest migration: patterns, phases and signals. Dev Biol. 2010[citado 14 Ene 2013];344:556-8.
40. Godfrey M, Vandermark N, Wang M. Prenatal diagnosis and a donor splice sie mutation in fibrillin in a family whit Marfan syndrome. Am J Hum Genet. 1993[citado 14 Ene 2013];53:472-80.
41. Lopes LM, Cha SC, De Moraes EA. Ecocardiographic diagnosis of fetal Marfansíndrome at 34 weeks gestation. PrenatDiagn. 1995[citado 14 Ene 2013];15:183-5.
42. Nyberg DA, McGahan JP, Pretorius DH. Diagnostic imaging of fetal anomalies. Philadelphia: Lippincott Williams and Wilkins; 2003.
43. Maldonado Martínez Y, Torres Molina A, Durán Lobaina D. Síndrome de Costello. Presentación de un caso. Medisur. 2014[citado 13 Ene 2013];12(2).
44. Tongsong T, Chanprapaph P. Prenatal sonographic diagnosis of Holt-Oram síndrome. J ClinUltrasound. 2010[citado 13 Ene 2013];28:98-100.
45. Kameda Y. Hoxa 3 and signaling molecules involved in aortic arch patterning and remodeling. Cell Tissue Rev. 2009[citado 14 Ene 2013];336:165-78.
46. Fyler DC. Coarctation of the aorta. In: Fyler DC, Ed NodasPediatr Cardiology. Philadelphia: Hanley and Belfus;1992:549.
47. Bader RS, Chitayar D, Kelly E. Fetal rhabdomyoma: prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex. J Pediatr. 2003[citado 14 Ene 2013];143:620-4.
Recibido: 7 de octubre de 2017.
Aprobado: 11 de noviembre de 2017.
Yamilet Corona Carnero. Departamento de Genética. Municipio Habana del Este. La Habana, Cuba.
Correo electrónico: yamilet.corona@infomed.sld.cu

