










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Salud Animal
versión impresa ISSN 0253-570X
Rev Salud Anim. v.29 n.1 La Habana ene.-abr. 2007
Artículo original
DISEÑO DE UN ENSAYO DE REACCIÓN EN CADENA DE LA POLIMERASA PARA LA DETECCIÓN DE Herpesvirus bovino tipo 1
DESIGN OF A POLYMERASE CHAIN REACTION ASSAY FOR THE DETECTION OF bovine Herpesvirus type 1
Majela Rodríguez, Mislay Avila, Heidy Díaz de Arce y Maritza Barrera
Centro Nacional de Sanidad Agropecuaria (CENSA), Apartado 10, San José de las Lajas, La Habana, Cuba. Correo electrónico: majela@censa.edu.cu
RESUMEN
Se describe el diseño de un ensayo de reacción en cadena de la polimerasa (PCR) para la detección de herpesvirus bovino tipo 1 (HVB-1). Este se basó en la amplificación de una porción del gen tk viral, para lo cual se ensayaron diferentes concentraciones de los cebadores, de cloruro de magnesio y diferentes temperaturas de alineamiento. Además, se evaluaron los parámetros de desempeño analítico, sensibilidad y especificidad, con resultados satisfactorios. Bajo las condiciones óptimas se logró la amplificación de la banda de 202 pb esperada y se demostró la utilidad del ensayo para la identificación de aislados.
Palabras clave: herpesvirus bovino tipo 1; diagnóstico; PCR.
ABSTRACT
The design of a Polymerase Chain Reaction (PCR) assay for the detection of bovine herpesvirus type 1 (BHV-1) was described. This one was based on the amplification of a portion of the viral tk gene, for which different concentrations of primers, magnesium chloride and different annealing temperatures were tried. In addition, the parameters of analytical performance, sensitivity and specificity were evaluated with satisfactory results. Under the optimal conditions, the amplification of the band 202 pb expected was obtained, and the utility of the test for the identification of isolates was demonstrated.
Key words: bovine herpesvirus type 1; diagnosis; PCR.
INTRODUCCIÓN
Herpesvirus bovino tipo 1 es el agente causal de la rinotraqueitis infecciosa bovina (IBR), una de las enfermedades que más incidencia tiene en los costos de producción de los bovinos (2). Además es el responsable de otros síndromes clínicos, tales como vulvovaginitis pustular infecciosa (en las hembras) o balanopostitis pustular infecciosa (en los machos), conjuntivitis, aborto, enteritis e infección sistémica generalizada (13, 1, 16, 11).
Una de sus características más importantes es que establece infección latente en las neuronas sensoriales del ganglio trigémino o sacro e, incluso, en las tonsilas (28, 12). Esto tiene una significativa importancia epizootiológica por el peligro que representan los animales sin manifestaciones clínicas y serológicamente negativos, pero que pueden liberar virus bajo condiciones de estrés y por tanto constituyen una fuente de infección permanente para otros animales.
En Cuba, el diagnóstico de laboratorio de HVB-1 se realiza mediante el uso de métodos clásicos como la inmunofluorescencia e inmunoperoxidasa directas, el aislamiento viral e identificación posterior por seroneutralización viral y por la detección de anticuerpos específicos en muestras de sueros pareados o simples. Las técnicas inmunohistoquímicas, aunque son más rápidas que el aislamiento viral se caracterizan por una menor sensibilidad y especificidad, requieren de la presencia del antígeno viral en la célula intacta, la calidad de la muestra repercute en los resultados y en ocasiones se basan en el uso de antisueros obtenidos contra cepas heterólogas, lo cual no asegura que se detecten determinadas variaciones antigénicas del aislado problema (18). El aislamiento viral, por su parte, requiere de facilidades de cultivo celular y mucho tiempo para la obtención de los resultados y las pruebas de seroconverción toman un mínimo de 14 a 21 días y pueden escapar de la detección, animales persistentemente infectados con bajos títulos de anticuerpos (15, 21).
Esto ha mostrado la necesidad de acudir a métodos de diagnóstico más rápidos y precisos que posibiliten tomar medidas efectivas para limitar la diseminación de la infección a otros animales, reducir el riesgo inherente a una enfermedad clínica no diagnosticada que pueda ser exótica o al uso indebido de antibióticos en enfermedades mal diagnosticadas. La PCR se ha utilizado con estos fines en la virología veterinaria de forma general (4). Específicamente en el diagnóstico de HVB-1, esta técnica ha mostrado ser una herramienta muy útil para la detección en semen (23, 7) y otras muestras clínicas (20, 25). La selección del gen tk para la PCR, ha sido muy efectiva debido a su importancia en la patogenisidad viral de todos los herpesvirus bovinos (26). Su secuencia fue por primera vez determinada por Bello et al. (5) y en la actualidad existen numerosas secuencias del mismo publicadas en bases de datos de acceso libre. Apoyado en estas premisas se propuso como objetivo del presente trabajo: diseñar un ensayo de PCR para la detección de HVB-1.
MATERIALES Y MÉTODOS
Virus y Células
Se trabajó con la suspensión viral obtenida mediante la propagación de la cepa de HVB-1, E8, aislada en Cuba por Noda et al. (19) en un brote de queratoconjuntivitis, a una multiplicidad de infección de 1 DICT50/célula en cultivos de la línea celular Madin-Darby Bovine Kidney (MDBK, ATCC CCL 22, certificada libre de virus de la diarrea viral bovina).
Extracción de ADN
Se procedió según lo descrito por Sambrook et al. (22) con algunas modificaciones.
Se trataron 200 mL de suspensión viral (105.45 DICT50/mL) con duodecil sulfato de sodio y proteinasa K a 1% y 0.2 mg/mL de concentración final, respectivamente, durante 1 hora a 56ºC en un baño serológico.
En paralelo se trataron 200 mL de un sobrenadante de células MDBK no infectadas como control negativo. Se mezclaron con 200 mL de fenol saturado con Tris 1M pH 8.0 y se centrifugaron 5 min a 12000 rpm (Centrífuga eppendorf 5415D). La fase acuosa se mezcló con 200 mL de cloroformo-isoamil alcohol (49:1 v/v) y se centrifugó a 12000 rpm durante 5 min. Se extrajo la fase superior, el ácido nucleico se precipitó mediante la adición de 2 volúmenes de etanol absoluto frío y 1/10 del volumen de acetato de sodio 3M pH 5.2 durante 2 horas a -80ºC y se centrifugó durante 15 min a 14000 rpm. Finalmente se lavó el precipitado con etanol al 70%, se secó al aire y se resuspendió en 20 mL de TE 1X. La concentración y pureza se determinaron mediante un espectrofotómetro Ultrospec® 2100 pro (Amersham Pharmacia Biotech) y se utilizaron cantidades entre 0.1 y 0.2 mg en el PCR.
Ensayo de PCR
Cebadores: Se sintetizaron dos oligonucleótidos nuevos, S21 5'GCGGGCCTGGTTGCGTACTAC3' y A21 5'AGCAGATCTTCCGCGTTGATC3' correspondientes a los nucleótidos 514-535 y 716-695, respectivamente, en el gen tk de HVB-1.1 (número de acceso al GenBank D00438 (17)).
Amplificación: Los volúmenes y concentraciones finales de la reacción de PCR que empleamos como base para la estandarización fueron las recomendadas por el boletín técnico Nº 254 de la PCR Core System de Promega®, donde se establecen las siguientes pautas: tampón de reacción de la Taq ADN polimerasa (50 mM KCl, 10 mM Tris-HCl pH 9.0 y 0.1% Triton® X-100), solución de cloruro de magnesio (0.5-3 mM), mezcla de nucleótidos (0.2 mM cada uno), cebadores (0.1-1.0 ìM), Taq ADN Polimerasa (1.25 U) y completar con agua hasta un volumen final de 50 ìL. Como control negativo de la reacción se sustituyó el ADN molde por el mismo volumen de agua libre de nucleasas (Promega).
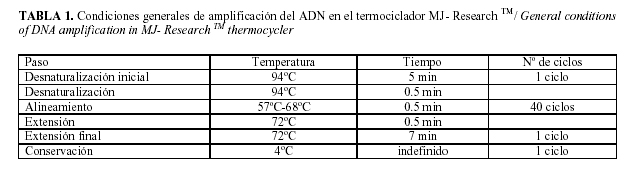
Las condiciones iniciales de amplificación del ADN en el termociclador MJ- Research TM, las cuales habían sido utilizadas con éxito en el laboratorio para otros cebadores, están descritas en la Tabla 1.
Algunas de las variables y condiciones de partida fueron estandarizadas y en todos los casos se realizó un mínimo de tres réplicas por ensayo.
Análisis del producto amplificado por PCR
El producto de PCR con 1X de tampón de carga azul-naranja (Promega) fue analizado por electroforesis en geles de agarosa al 2% teñido con bromuro de etidio 0,08 mg/mL. La corrida se realizó en tampón TBE 0.5X a un voltaje constante de 100 V por 30 min. Para determinar la talla aproximada del producto fue incluido un patrón de peso molecular 100pb DNA Ladder (Promega) con un rango de tallas de 100-1500pb. Las bandas fueron visualizadas a una longitud de onda de 312 nm y se fotografiaron con cámara digital (Hewlett Packard C6324 A) para recoger los resultados. Se tuvo en cuenta la intensidad de la banda de amplificación para seleccionar las condiciones óptimas.
Determinación de la especificidad analítica de los cebadores
La especificidad analítica se determinó mediante la evaluación de diferentes ADN moldes provenientes de:
- Cepa de HVB-2 donada gentilmente por el DrC. A. Bartha, Academia de Ciencias Veterinarias de Hungría.
- Cepa Misk/67 de adenovirus bovino 8 y cepa Sofía de adenovirus bovino 9 donadas gentilmente por Instituto de Phylaxia, Hungría.
- Cepa del virus de la enfermedad de Aujeszky: V208, aislada en Cuba por Díaz et al. (9).
Determinación del límite de detección del ensayo
Con el propósito de determinar el límite de detección del ensayo se realizó una curva de diluciones de punto final en medio de cultivo celular, de la suspensión viral de E8. A partir de estas diluciones se aisló el ADN por el método antes descrito y se utilizó como molde para el PCR donde se evaluaron valores de 1, 5 y 8% de DMSO en la mezcla de reacción.
Identificación de aislados
Los aislados fueron obtenidos previamente a partir de exudados nasales de seis toros con signos clínicos compatibles a la IBR y cuatro vacas utilizadas como controles negativos. La identificación se realizó con la PCR diseñada y mediante la inmunofluorescencia directa (IFD) con IgG de bovino contra la cepa E8, conjugado con isotiocianato de fluoresceína (1:16): obtenido en el Laboratorio de Virología Animal, Centro Nacional de Sanidad Agropecuaria, según Delgado et al. (8) y Barrera (3).
RESULTADOS Y DISCUSIÓN
Con el método de extracción ensayado se obtuvieron valores de concentración de ADN total entre 50 y 70 mg/mL y la pureza, con relación a la presencia de proteínas contaminantes, se mantuvo por encima de 2 en todos los casos, lo que se corresponde con un elevado grado de pureza según Sambrook et al. (22). La calidad del material genético obtenido resultó adecuada para el ensayo de PCR mediante el cual se obtuvo un producto amplificado de 202 pb, talla esperada según el diseño de los cebadores (Fig. 1).

El hecho de que el genoma de los herpesvirus sea rico en GC (32-75%) (10), se tuvo en cuenta para el diseño de cebadores que permitieron usar un rango de temperaturas de alineamiento elevadas. Al evaluar las temperaturas 57, 60, 62 y 65°C se obtuvieron bandas de amplificación de similar intensidad (Fig. 2). Con la temperatura de alineamiento de 68°C, se obtuvo una banda de reducida intensidad comparada con el resto. Esto se debe a que elevadas temperaturas disminuyen la sensibilidad de la reacción (29). Se seleccionó la temperatura de 65°C porque se conoce que valores elevados minimizan las uniones inespecíficas de los cebadores y por tanto incrementan la especificidad de la reacción (29), lográndose el mejor balance entre sensibilidad y especificidad de unión de los cebadores.
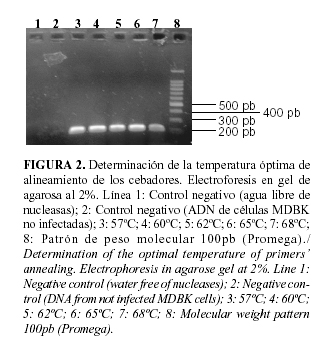
Para seleccionar la concentración óptima de cebadores en un ensayo de PCR, se recomienda evaluar valores en un rango entre 0.1-1mM (22). La concentración óptima obtenida en este trabajo fue de 0.6 mM, la cual se encuentra en el rango de las recomendadas por la literatura. Las concentraciones más bajas resultaron en poco producto de PCR, de forma similar ocurrió a concentraciones elevadas (Fig. 3).
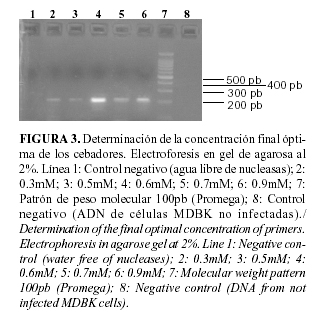
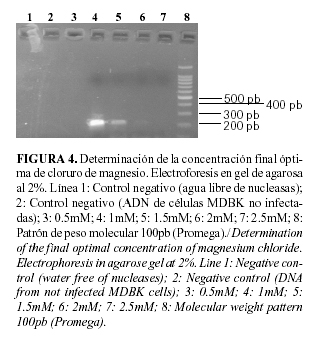
Algunos cebadores funcionan de manera similar a varias concentraciones de Mg2+ mientras que otros pueden tener requerimientos muy específicos. La pareja de cebadores diseñada se ajusta al segundo caso, ya que a concentraciones superiores a 1mM de cloruro de magnesio, disminuyó considerablemente la intensidad de la banda y a 0.5 mM no se obtuvo amplificación, por lo que el valor óptimo fue 1mM (Fig. 4).
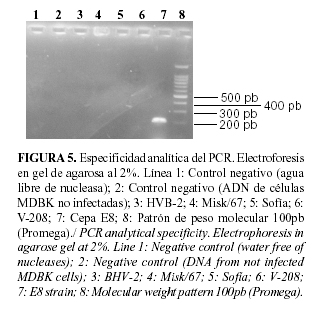
En la Figura 5, se muestra la especificidad de los cebadores para detectar HVB-1. No ocurrió amplificación al evaluar herpesvirus heterólogos como HVB-2 o el virus de la enfermedad de Aujeszky u otros virus bovinos de genoma ADN que pudieran estar presentes en muestras de exudados oculares o nasales como los adenovirus 8 y 9. Sólo se obtuvo producto amplificado para la cepa E8, utilizada como control positivo de la reacción, lo cual indica que el diseño de los cebadores es adecuado y que solamente identifican la región del gen tk de interés para el diagnóstico.
El límite de detección del ensayo de PCR fue de 2x102.45 DICT50/mL, el cual mejoró ligeramente hasta 102.45 DICT50/mL al añadir 5% de DMSO en la mezcla de reacción (Fig. 6). Este reactivo ha sido muy utilizado en el PCR de genomas con elevados contenidos de GC y estructuras secundarias que pueden inhibir el alineamiento y extensión de los cebadores (30 y 6).

La literatura informa que durante 7 días postinfección se elimina gran cantidad de partículas virales por la secreción nasal (24), 107.75 DICT50/g de mucosa, por lo que el límite de detección obtenido, pudiera ser suficiente para detectar HVB-1 en muestras clínicas.
Este ensayo fue capaz de detectar el ácido nucleico viral en todos los aislados provenientes de animales con signos clínicos compatibles con la IBR y el resto resultó negativo. Esto evidencia una mayor sensibilidad de la PCR desarrollada respecto a la IFD que sólo detectó dos de las muestras presuntamente positivas (Tabla 2).
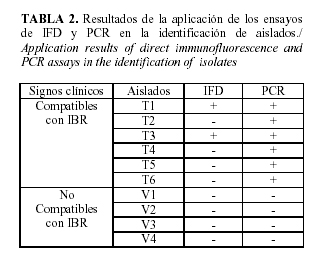
El comportamiento de este parámetro concuerda con lo planteado por Wiedman et al. (27); Yason et al. (30) y Moore et al. (18) y se debe a que la PCR está basada en la detección del ácido nucleico (4), no requiere que la muestra tenga una calidad óptima y es capaz de detectar virus en animales infectados latentemente (14).
Los resultados obtenidos confirman la utilidad del ensayo de PCR para la identificación de aislados, el cual se empleará además, en el diagnóstico de HVB-1 a partir de muestras clínicas (exudados nasales y oculares, semen importado, etc.) y para descartar la presencia de este agente en el esquema de diagnóstico de las enfermedades vesiculares. Esto permitirá la liberación de la cuarentena de los animales o que se tomen las medidas de control adecuadas en el menor tiempo posible y de esa forma evitar la diseminación de la enfermedad y contribuir a la disminución de las pérdidas económicas.
REFERENCIAS
1. Abinanti FR, Plumer GH. The isolation of IBR virus from cattle affected with conjunctivitis, observations on the experimental infection. Am J Vet Res. 1961;22:13-17.
2. Armstrong JA, Pereira HG, Andrews CH. Observation of the virus of IBR and its affinity with the Herpesvirus group. Virology 1961;14:276-285.
3. Barrera M. Herpesvirus Bovino-1: Obtención de Medios de Diagnóstico y Prevención. Tesis para la Opción del Grado de Científico de Dr.C. Veterinaria. ISCAH. CENSA. Cuba. 1996.
4. Belak S, Ballagi-Pordany A. Application of the polymerase chain reaction (PCR) in veterinary diagnostic virology. Vet Res Commun. 1993;17:55-72.
5. Bello LJ, Whitbeck JC, Lawrence WC. Sequence and transcript analysis of the bovine herpesvirus 1 Thymidine kinase locus. Virology 1992;189:407-414.
6. Cândido AL, Bontempo ED, Resende M. Polymerase Chain Reaction of Genomic Fragments of Bovine Herpesvirus-1. Mem Inst Oswaldo Cruz 2000;95(2):225-226.
7. Deka D, Ramneek Maiti NK, Oberoi MS. Detection of bovine herpesvirus-1 infection in breeding bull semen by virus isolation and polymerase chain reaction. Rev sci tech Off Int Epiz. 2005;24(3):1085-1094.
8. Delgado I, Barrera M, Tuero C, Rodríguez N. Comparación de tres métodos de detección de antígeno para el diagnóstico de HVB-1. Rev Salud Anim. 1992;14:143-148.
9. Díaz JCD, Castell S, Barrera M. Diagnosis of Aujeszky's disease virus infection in samples of doubtful etiology. Rev Salud Anim. 2006;28(1):36-39.
10.Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. (eds) Virus Taxonomy, VIIIth Report of the ICTV.2005 Elsevier, Academic Press. <http://www.ncbi.nlm.nih.gov/ICTVdb/Ictv/fr-fst-g.htm> Última actualización enero del 2005.
11.Gibbs EPJ, Rweyemamu MM. Bovine herpesviruses. Part I. Bovine herpesvirus 1. Vet Bull. 1977;47:317-343.
12.Jones C. Herpes simplex Virus Type 1 and Bovine Herpesvirus 1 Latency. Clin Microbiol Rev. 2003;16:79-95.
13.Kendrick JW, Gillespie JH, McEntee K. Infectious pustular vulvovaginitis of cattle. Cornell Vet. 1958;48:458-495.
14.Keuser V, Schynts F, Detry B, Collard A, Robert B, Vanderplasschen A. Improved Antigenic Methods for Differential Diagnosis of Bovine, Caprine, and Cervine Alphaherpesviruses Related to Bovine Herpesvirus 1. J Clin Microbiol. 2004;42(3):1228-1235.
15.Kirkbride CA. Etiologie agents in a 10 year study of bovine abortions and stillbirths. J Vet Diagn Investig. 1992;4:160-175.
16.McKercher DG, Wada EM. The virus of IBR cause of abortion in cattle. J Amer Vet Med Ass. 1964;144:136-158.
17.Mittal SK, Field HJ. Analysis of bovine herpesvirus type 1 thymidine kinase (tk) gene from wild-type virus and tk-deficient mutants. J Gen Virol. 1989;70:901-918.
18.Moore S, Gunn M, Walls D. A rapid and sensitive PCR-based diagnostic assay to detect bovine herpesvirus 1 in routine diagnostic submissions. Vet Microbiol. 2000;75:145-153.
19.Noda J, Nuñez A, García J. Aislamiento del virus de la rinotraqueitis infecciosa bovina en terneros con queratoconjutivitis. Rev Salud Anim. 1984;6:651-653.
20.Pompeo MC, Alfieri AF, Valadares AFF, Rezler SW, Médici KC, Alfieri AA. Rapid detection and differentiation of bovine herpesvirus 1 and 5 glycoprotein C gene in clinical specimens by multiplex-PCR. J Virol Methods 2005;128:183-188.
21.Rola J, Larska M, Polak MP. Detection of bovine herpesvirus 1 from an outbreak of infectious bovine rhinotracheitis. Bull Vet Inst Pulawy. 2005;49:267-271.
22.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press. 1989.
23.Smits CB, Van Maanen C, Glas RD, De Gee ALW, Dijkstrab T, Van Oirschot JT et al. Comparison of three polymerase chain reaction methods for routine detection of bovine herpesvirus 1 DNA in fresh bull semen. J Virol Methods. 2000;85:65-73.
24.Straub OC. Agentes víricos como causa de trastornos en la reproducción bovina. MV Rev Cien Vet. 1994;10(2):15-21.
25.Takiuchi E, Médici KC, Alfieri AF, Alfieri AA. Bovine herpesvirus type 1 abortions detected by a semi-nested PCR in Brazilian cattle herds. Res Vet Sci. 2005;79:85-88.
26.Tikoo SK, Campos M, Babiuk LA. Bovine herpesvirus 1 (BHV-1): Biology, pathogenesis, and control. Adv Virus Res. 1995;45:191-223.
27.Wiedman M, Brandon R, Wagner P, Dubovi E, Batt CA. Detection of bovine herpesvirus-1 in bovine semen by a nested PCR. J Virol Meth.1993;44:129-140.
28.Winkler M, Doster A, Jones C. Persistence and Reactivation of Bovine Herpesvirus 1 in the Tonsils of Latently Infectes Calves. J Virol. 2000;74:5337-5346.
29.Wu DY, Ugozzoli L, Pal BK, Qian J, Wallace B. The effect of temperature and oligonucleotide primer length on the specificity and efficiency of amplification by the polymerase chain reaction. DNA and Cell Biology 1991;10:233-238.
30.Yason CV, Harris LM, McKenna PK, Wadowska D, Kibenge FSB. Establishment of Conditions for the Detection of Bovine Herpesvirus-1 by Polymerase Chain Reaction Using Primers in the Thymidine Kinase Region. Can J Vet Res. 1995;59:94-101.
(Recibido 29-9-2006; Aceptado 29-12-2006)


{kind=link}