










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Salud Animal
versión impresa ISSN 0253-570X
Rev Salud Anim. v.33 n.1 La Habana ene.-abr. 2011
Artículo original
EXPRESIÓN in vitro EN Escherichia coli, DE UN FRAGMENTO DEL GEN vlhA.5.02 DE LA PRINCIPAL FAMILIA DE HEMAGLUTININAS DE Mycoplasma gallisepticum
In vitro EXPRESSION IN Escherichia coli OF THE vlhA.5.02 GENE FRAGMENT, A MEMBER OF THE MAJOR HEMAGLUTININ FAMILY OF Mycoplasma gallisepticum
J.A. Agüero*, H. Hotzel**, Lisandra Aguilar*, Claudia Lima*, K. Sachse**, Siomara Martínez*
*Centro Nacional de Sanidad Agropecuaria (CENSA). Carretera de Jamaica y Autopista Nacional. Apartado 10. San José de las Lajas. La Habana, Cuba. Correo electrónico: jaaguero@censa.edu.cu; **Instituto Friedrich-Loeffler, Instituto de Patogénesis Molecular, Naumburger Str. 96ª, Jena, Alemania
RESUMEN
Mycoplasma gallisepticum es el agente etiológico fundamental del síndrome respiratorio crónico de los pollos; constituye el micoplasma más importante desde el punto de vista económico para la avicultura. Un fragmento de ADN de aproximadamente 800pb de este microorganismo, amplificado mediante la reacción en cadena de la polimerasa a partir del plásmido pTTQ18 recombinante, se clonó en el vector de expresión pQE-32. El polipéptido se expresó fusionado a una secuencia de seis histidinas, por lo que la talla final fue de 31kDa, y se detectó mediante inmunotransferencia con un antisuero policlonal contra M. gallisepticum y con un anticuerpo monoclonal contra histidina. El polipéptido se purificó mediante cromatografía de afinidad con quelatos metálicos níquel-ácido nitrilotriacético, lográndose aproximadamente un 90% de pureza.
Palabras clave: Mycoplasma gallisepticum; clonaje, expresión; purificación; PCR; cromatografía de afinidad por quelatos metálicos níquel-ácido nitrilotriacético (Ni-NTA).
ABSTRACT
Mycoplasma gallisepticum, the causal agent of the chronic respiratory disease of chickens, is the most economically significant for poultry industry. An 800pb DNA fragment of Mycoplasma gallisepticum which was amplified from the recombinant vector pTTQ18 was cloned into the expression vector pQE-32. The polypeptide was expressed as a fusion protein to 6X-histidine tag; hence its final size was 31kDa. It was detected by Western blott being recognized by a polyclonal antiserum to M. gallisepticum, as well as by a monoclonal antibody to histidine. The polypeptide was purified by nickel-nitrilotriacetic acid (Ni-NTA) metal-affinity cromatography yielding a final product of nearly 90% of purity.
Key words: Mycoplasma gallisepticum; cloning; expression; purification; PCR; nickel-nitrilotriacetic acid (Ni-NTA) metal-affinity cromatography.
INTRODUCCIÓN
Mycoplasma gallisepticum fue aislado por primera vez en 1935 y es el principal agente causal de la enfermedad respiratoria crónica de los pollos e infecciones de sinusitis en pavos (1); constituye, además, la especie de mayor importancia económica dentro de los micoplasmas aviares. Aunque esta enfermedad no necesariamente provoca elevada mortalidad, la morbilidad es alta, y los consiguientes efectos perjudiciales de la enfermedad son debido a la disminución en la producción de huevos, retraso del crecimiento y predisposición a las infecciones secundarias virales y bacterianas (2,3). De esta manera las parvadas de gallinas reproductoras pueden quedar infectadas, lo que provocaría la pérdida del mercado de exportación de su progenie o incluso podría obligar al sacrificio de las mismas (4).
El esclarecimiento de los mecanismos patogénicos por los cuales M. gallisepticum ejerce sus efectos sobre las aves de corral, es crítica en la búsqueda racional de mejores vacunas y estrategias de control. Hasta la fecha se han identificado algunos determinantes de virulencia o determinantes asociados a la virulencia. Dentro de estos tenemos, con alguna importancia, moléculas involucradas en vías metabólicas como la dihidrolipoamida deshidrogenasa (LPD), un componente del complejo piruvato deshidrogenasa, que se requiere en el crecimiento in vivo y la supervivencia en el hospedero (5). Por otra parte, y como piezas fundamentales en la virulencia de esta bacteria están las citadhesinas. La adhesión de M. gallisepticum a las células del epitelio respiratorio constituye un paso crítico para la infección y la enfermedad (6), y se logra mediante las lipoproteínas localizadas en su superficie. Se han reportado varias de estas proteínas como son la familia de citadhesinas-hemaglutininas VlhA; otras citadhesinas como GapA (7,8); proteína semejante a OsmC, MG1142 (9); PlpA (10) y PvpA (11), y proteínas accesorias a las citadhesinas, como CrmA (7,12).
La obtención de antígenos vía ADN recombinante es una de las alternativas utilizadas con el objetivo de estudiar su función en los mecanismos de patogenicidad y evasión de la respuesta inmune por parte del microorganismo. Saito et al. obtuvieron en 1993 un polipéptido recombinante de 29kDa procedente de M. gallisepticum (1). El mismo es un fragmento de VlhA5.02, uno de los integrantes de la familia de hemaglutininas VlhA (antiguamente llamada pMGA y denominada así después de la secuenciación del genoma de este microorganismo) (13). Las numerosas lipoproteínas codificadas por esta familia de genes, media la variación de la arquitectura de la superficie de M. gallisepticum y se cree que está involucrada en la evasión del sistema inmune del hospedero (14). Este polipéptido protegió parcialmente a pollos confrontados con una cepa virulenta de M. gallisepticum. Los anticuerpos obtenidos contra este polipéptido reconocen a tres proteínas de aproximadamente 29, 40 y 65kDa de lisados celulares de M. gallisepticum y sueros contra este microorganismo reaccionan con esta proteína recombinante (1).
Estos resultados sugieren que un preparado vacunal enriquecido con esta proteína, puede elevar sus niveles de protección contra una infección de M. gallisepticum. Teniendo en cuenta todo lo anterior, en nuestro laboratorio, a partir de la secuencia reportada por Saito et al. (1), se clonó y expresó el gen que codifica para este polipéptido en tres vectores de expresión: pTTQ18, pRIV2 y pTrcHis (15). A pesar de que se logró la expresión de la proteína en los tres casos, los niveles de expresión no fueron suficientes para realizar la purificación y posteriormente los estudios in vitro.
El objetivo de este trabajo fue realizar el clonaje, evaluar diferentes sistemas de expresión y purificar un fragmento de la hemaglutinina VlhA5.02 de Mycoplasma gallisepticum.
MATERIALES Y MÉTODOS
Amplificación de un fragmento de 800pb de la proteína VlhA5.02 de Mycoplasma gallisepticum. La secuencia de 800pb que codifica para un polipéptido de 29kDa de la hemaglutinina VlhA5.02 de M. gallisepticum se amplificó mediante reacción en cadena de la polimerasa (PCR, del inglés polymerase chain reaction) a partir del plásmido pTTQ18 recombinante (15) con la enzima Pwo (Boehringer Mannheim) siguiendo el protocolo descrito por Chávez y col. (16). El producto de PCR se chequeó por electroforesis en gel de agarosa al 0,8% (P/V).
Construcción del plásmido recombinante pKJY29. El plásmido recombinante pKJY29 se construyó mediante la inserción del producto de PCR obtenido anteriormente, en los sitios SmaI y HindIII del vector pQE-32 (QIAGEN). La ligazón de 100ng de vector con 116ng del inserto se realizó a 14°C durante toda la noche en presencia de ADN T4 ligasa (Promega), solución tampón de ADN T4 ligasa 10x (Tris 1M pH 7.5, MgCl2 1M), ditiotreitol 5mM, ATP 10mM, en un volumen final de 20 mL.
Obtención de cepas recombinantes. Se transformaron 200mL de células competentes de la cepa M15 de Escherichia coli por electroporación con 5mL del producto de la ligazón. Se aplicaron 100mL de la mezcla de transformación en placas de medio sólido Luria-Bertani (LB) suplementado con ampicillina (100mg/mL) y kanamicina (25mg/mL) y se incubaron a 37°C durante toda la noche. Las posibles colonias recombinantes se monitorearon por PCR, para el cual se emplearon los mismos cebadores usados en la amplificación inicial del fragmento de VlhA5.02. El producto de PCR se chequeó por electroforesis en gel de agarosa al 0,8% (P/V).
Expresión mediante inducción con IPTG y purificación del polipéptido recombinante. Se realizó la expresión de las colonias recombinantes mediante inducción con isopropil-b-D-tiogalactosidasa (IPTG). Se preparó 1L de cultivo LB suplementado con ampicillina (100mg/mL) y kanamicina (25mg/mL), y cuando este alcanzó una DO600 de 0.6, se indujo la expresión con IPTG 1mM. Para realizar el experimento de purificación se tomó la colonia que presentó un mayor nivel de expresión de la proteína recombinante. La purificación se llevó a cabo por cromatografía de afinidad por quelatos metálicos níquel-ácido nitrilotriacético (Ni-NTA) según las instrucciones del fabricante (QIAexpress, QIAGEN). El cultivo se centrifugó y el precipitado se resuspendió en tampón de lisis (NaH2PO4 50mM, pH8; NaCl 300mM; imidazol 10mM) y la ruptura se realizó mediante sonicación en presencia de lisozima (1mg/mL). La muestra se centrifugó durante 30 min a 13 000 x g, el sobrenadante se aplicó en la columna de Ni-NTA, la cual se lavó con solución tampón (NaH2PO4 50mM, pH8; NaCl 300mM; imidazol 20mM) y la proteína se eluyó con tampón de elución (NaH2PO4 50mM, pH8; NaCl 300mM; imidazol 250mM). La purificación en su totalidad se realizó por gravedad.
Expresión del polipéptido recombinante mediante autoinducción con lactosa. Con el objetivo de abaratar los costos de la obtención del péptido recombinante, así como para facilitar la misma, se estudió la expresión del fragmento recombinante de VlhA5.02 por el método de autoinducción desarrollado por Studier (17) con algunas modificaciones, el cual había sido empleado anteriormente con éxito en nuestro laboratorio (18). La cepa BL21(DE3)pLys de E. coli se transformó con la construcción recombinante pKJY29 mediante electroporación, y el producto de la transformación se plaqueó en LB sólido suplementado con ampicillina (100mg/mL) y cloranfenicol (35mg/mL). Las colonias recombinantes crecidas se cultivaron toda la noche a 37°C en medio líquido LB suplementado con ampicillina (100mg/mL). Al día siguiente, con 10mL de este cultivo se inocularon 100mL (en un erlenmeyer de 1L) de medio de auto-inducción (triptona 1%, extracto de levadura 0.5%, Na2HPO4 25mM; KH2PO4 25mM; NH4Cl 50mM; Na2SO4 5mM; glicerol 0.5%; glucosa 0.05%; alfa-lactosa monohidratada 0.2%; MgSO4 7H2O 2mM; FeCl3 0.05mM) suplementado con ampicillina (100ìg/mL) y cloranfenicol (35mg/mL), y se cultivó por 16 horas a 37°C con agitación a 310rpm. Se recolectaron las células por centrifugación a 5 000 x g por 10min y se conservaron a -20°C.
Análisis del polipéptido expresado mediante SDS-PAGE e inmunotransferencia. Tanto los niveles de expresión de la proteína recombinante como su purificación se chequearon por electroforesis en gel de poliacrilamida (SDS-PAGE) al 10%, y los geles fueron examinados con el programa de análisis densitométrico Lane Manager (TDI) para cuantificar la proteína. Para la identificación de la proteína, esta se inmovilizó en membrana de nitrocelulosa y la detección se realizó con un antisuero hiperinmune contra la cepa PG31 de M. gallisepticum y con un anticuerpo monoclonal anti-histidina (QIAGEN). Se empleó un sistema quimioluminiscente (ECL, Amersham) para el revelado de las reacciones.
RESULTADOS Y DISCUSIÓN
Selección del vector y las cepas de expresión. La expresión de proteínas recombinantes clonadas en los vectores pQE es inducida tanto en presencia de IPTG como de la alolactosa, análogos de la lactosa que se unen al represor lac y lo inactivan. De ahí que se escogiera el plásmido pQE-32 para los dos experimentos de expresión: inducción y autoinducción. Este plásmido contiene además una secuencia que codifica para seis histidinas en el extremo 5' del sitio múltiple de clonaje, que se expresa fusionada al extremo N-terminal de la proteína clonada, lo que facilita la unión de los productos de expresión a agarosa modificada con grupos Ni-NTA y, por ende, su posterior purificación. Esta región de seis histidinas es poco inmunogénica y en la mayoría de los casos no interfiere en la estructura y función de las proteínas purificadas, lo que ha sido demostrado en una amplia variedad de proteínas que incluyen enzimas, factores de transcripción y vacunas.
La cepa M15 de E. coli empleada en este sistema (QIAexpress, QIAGEN) contiene el plásmido pREP4 de bajo número de copias, el cual confiere resistencia a kanamicina, codifica para el gen lacI, expresando constitutivamente el represor Lac. La cepa BL21(DE3)pLysS de E. coli, empleada en el experimento de autoinducción, contiene un plásmido que expresa constitutivamente la lisozima T7, enzima que inhibe la T7 ADN polimerasa empleada en estos sistemas, lo que permite no se expresen niveles basales de proteína, y además esta enzima facilita la ruptura celular, lo cual es importante antes de la purificación de la proteína.
Clonaje del fragmento de ADN del gen vlhA 5.02 de Mycoplasma gallisepticum. Se decidió amplificar la secuencia del fragmento del gen vlhA5.02 de M. gallisepticum a partir del plásmido pTTQ18 recombinante donde se había clonado anteriormente, ya que dicho clonaje se comprobó por secuenciación y se demostró que el fragmento de interés no presentaba ningún error en su secuencia (15).
En el PCR realizado para obtener el fragmento de 800pb, se empleó una polimerasa de alta fidelidad de copia para garantizar que no hubiera errores de cambio de bases en el producto de reacción. Los cebadores empleados introducían en la secuencia dos sitios de reconocimiento para las enzimas de restricción EcoRI y HindIII. El plásmido hospedero pQE-32 posee sitio HindIII pero no sitio EcoRI, por lo que se utilizó el sitio SmaI del mismo y el extremo EcoRI de la secuencia se dejó intacto sin digerir y solo se digirió el HindIII. La presencia de un sitio romo y otro cohesivo garantizó que el fragmento se clonara en la dirección correcta.
El monitoreo por PCR de las colonias obtenidas en la transformación de las células hospederas demostró la presencia de cinco colonias recombinantes en las cuales se amplificó el fragmento de 800pb correspondiente al gen de interés.
Expresión con IPTG. A estas colonias se les evaluó la capacidad de expresión del polipéptido recombinante. Se observó la expresión de una proteína con una talla aparente de 31kDa que no aparecía en los controles negativos (Fig. 1). Esta proteína fue reconocida por un antisuero policlonal contra M. gallisepticum y por un anticuerpo monoclonal contra histidina (resultados no mostrados), lo que demostró que era la proteína esperada. La variación de la talla de la proteína de 29 a 31kDa se debe a la inclusión de la región de seis residuos de histidina, lo cual ya se había predicho mediante el análisis de la secuencia con el programa Vector NTI Advance 10.0. Los niveles de expresión alcanzados (~20%) fueron superiores a los logrados en los tres sistemas anteriores donde había sido clonado el polipéptido (15).
Purificación de la proteína recombinante. La purificación se desarrolló por afinidad con quelatos metálicos empleándose una matriz de agarosa con iones de níquel acoplados y el chequeo de la pureza del producto final se realizó por SDS-PAGE (Fig. 1). Se alcanzó un grado de pureza de aproximadamente un 90% (estimado por densitometría) en un solo paso de purificación. Este resultado es susceptible de ser mejorado en estudios posteriores; también se debe optimizar el proceso de purificación para aumentar los rendimientos de proteína por litro de cultivo, ya que en nuestros experimentos, en los cuales se obtuvieron masas bacterianas de niveles normales, solo se alcanzó 1mg/L y según lo reportado en la literatura con este sistema es posible obtener hasta 50mg/L. La causa de este bajo rendimiento pudiera ser que la proteína se expresa de forma insoluble y al centrifugar el lisado esta precipita y no permanece en el sobrenadante, por lo se pierde parte de la misma. Para evitar este problema sería necesario realizar la purificación en condiciones desnaturalizantes por ejemplo en presencia de urea. Otro fenómeno que también se ha observado cuando se purifican proteínas por este método, es que luego que se fijan a la columna, no eluyen de la misma si el proceso no se lleva a cabo en condiciones desnaturalizantes. Por todo esto sería importante estudiar posteriormente este fenómeno aplicado a nuestra proteína.
Es importante destacar que luego del proceso de purificación la proteína conservó sus propiedades antigénicas, ya que fue reconocida por el antisuero contra M. gallisepticum (Fig. 2A); además mostró reacción con el anticuerpo monoclonal contra histidina (Fig. 2B).
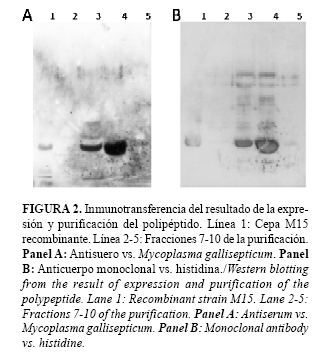
Expresión por autoinducción. Los resultados obtenidos mediante la autoinducción (Fig. 3) fueron similares a los obtenidos con el método tradicional de inducción con IPTG en cuanto a los niveles de expresión, aunque los rendimientos totales fueron superiores (aproximadamente 3 ó 4 veces) debido al mayor crecimiento de la masa celular que se genera en este caso (resultados no mostrados).

En resumen, se puede concluir que el antígeno recombinante que se obtuvo en este trabajo conserva sus propiedades antigénicas aún después del proceso de purificación, y que tanto el vector pQE-32 como la cepa hospedera BL21(DE3)pLys de E. coli, representan un sistema estable y fácilmente manejable para la expresión del fragmento de la proteína VlhA5.02 de M. gallisepticum.
REFERENCIAS
1. Saito S, Fujisawa A, Ohkawa S, Nishimura N, Abe T, Kodama K, et al. Cloning and DNA sequence of a 29 kilodalton polypeptide gene of Mycoplasma gallisepticum as a possible protective antigen. Vaccine. 1993;11(10):1061-1066.
2. Kleven SH. Mycoplasmas in the etiology of multifactorial respiratory disease. Poult Sci. 1998;77(8):1146-1149.
3. Ley DH, Berkhoff JE, Levisohn S. Molecular epidemiologic investigations of Mycoplasma gallisepticum conjunctivitis in songbirds by random amplified polymorphic DNA analyses. Emerg Infect Dis. 1997;3(3):375-380.
4. Bradbury JM. Gordon Memorial Lecture. Poultry mycoplasmas: sophisticated pathogens in simple guise. Br Poult Sci. 2005;46(2):125-136.
5. Hudson P, Gorton TS, Papazisi L, Cecchini K, Frasca S, Jr., Geary SJ. Identification of a virulence-associated determinant, dihydrolipoamide dehydrogenase (lpd), in Mycoplasma gallisepticum through in vivo screening of transposon mutants. Infect Immun. 2006;74(2):931-939.
6. Levisohn S. Early stages in the interaction between Mycoplasma gallisepticum and the chick trachea, as related to pathogenicity and immunogenicity. J Med Sci. 1984;20(10):982-984.
7. Papazisi L, Frasca S Jr, Gladd M, Liao X, Yogev D, Geary SJ. GapA and CrmA coexpression is essential for Mycoplasma gallisepticum cytadherence and virulence. Infect Immun. 2002;70(12):6839-6845.
8. Goh MS, Gorton TS, Forsyth MH, Troy KE, Geary SJ. Molecular and biochemical analysis of a 105 kDa Mycoplasma gallisepticum cytadhesin (GapA). Microbiology. 1998;144(11):2971-2978.
9. Jenkins C, Geary SJ, Gladd M, Djordjevic SP. The Mycoplasma gallisepticum OsmC-like protein MG1142 resides on the cell surface and binds heparin. Microbiology. 2007;153(5):1455-1463.
10.May M, Papazisi L, Gorton TS, Geary SJ. Identification of fibronectin-binding proteins in Mycoplasma gallisepticum strain R. Infect Immun. 2006;74(3):1777-1785.
11.Boguslavsky S, Menaker D, Lysnyansky I, Liu T, Levisohn S, Rosengarten R, et al. Molecular characterization of the Mycoplasma gallisepticum pvpA gene which encodes a putative variable cytadhesin protein. Infect Immun. 2000;68(7):3956-3964.
12.Papazisi L, Troy KE, Gorton TS, Liao X, Geary SJ. Analysis of cytadherence-deficient, GapA-negative Mycoplasma gallisepticum strain R. Infect Immun. 2000;68(12):6643-6649.
13.Papazisi L, Gorton TS, Kutish G, Markham PF, Browning GF, Nguyen DK, et al. The complete genome sequence of the avian pathogen Mycoplasma gallisepticum strain R(low). Microbiology. 2003;149(9):2307-2316.
14.Bearson SM, Collier SD, Bearson BL, Branton SL. Induction of a mycoplasma gallisepticum pMGA gene in the chicken tracheal ring organ culture model. Avian Dis. 2003;47(3):745-749.
15.Chávez YR, Fernández A, Martínez S. Clonaje y expresión de un gen que codifica un polipéptido de 29kDa de Mycoplasma gallisepticum. Rev Salud Anim. 1997;19(1):19-22.
16.Chávez YR, Fernández A, Martínez S. Amplificación in vitro de la secuencia de ADN de un polipéptido de 29 kDa de Mycoplasma gallisepticum. Rev Salud Anim. 1996;18(3):155-158.
17.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207-34.
18.Agüero JA, Sánchez O, Barrera M, Toledo JR. Molecular cloning and expression of a fragment of the gene codifying for the protein Erns of the Classical Swine Fever Virus. Rev Salud Anim. 2008;30(2):85-92.
(Recibido 3-12-2010; Aceptado 4-2-2011)

