










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Salud Animal
versión impresa ISSN 0253-570Xversión On-line ISSN 2224-4700
Rev Salud Anim. vol.37 no.1 La Habana ene.-abr. 2015
ORIGINAL ARTICLE
Molecular cloning and expression of a computationally predicted surface antigen of Mycoplasma gallisepticum
Clonaje molecular y expresión de un antígeno de superficie de Mycoplasma gallisepticum, predicho computacionalmente
José Antonio Agüero*, Lisandra Aguilar-Bultet, Ariadna Rodríguez
Microbial Genomics and Bioinformatics Laboratory. National Centre for Animal and Plant Health (CENSA), Apartado 10, San José de las Lajas, Mayabeque, CP 32700. Cuba.
ABSTRACT
Mycoplasmas, the simplest self-replicating organisms known, are distinguished phenotypically from other bacteria by their minute size and total lack of cell wall. The poultry industry is affected by several species of mycoplasmas, but Mycoplasma gallisepticum (MG) is the most economically significant one. The attachment of mycoplasmas to host respiratory epithelial cells constitutes a critical step in the pathway leading to infection and disease and is achieved by lipoproteins localized on the bacterial surface. In a recent in silico study, it was predicted a set of MG putative surface proteins with potential antigenic properties that could be used as candidates for exploring new vaccines, and for diagnostic tests as well. One of those potential candidates was the OppA. In this work we described the molecular cloning of the DNA segment that encodes a fragment of OppA, a polypeptide of 233 aa and about 30 kDa, as well as its expression, purification and immunogenic response induced by the product obtained.
Key words: Mycoplasma gallisepticum, surface protein, recombinant protein, IMAC purification.
RESUMEN
Los micoplasmas son los organismos autoreplicativos más simples que se conocen, y que se distinguen de otras bacterias por su pequeña talla y la total carencia de pared celular. La industria avícola está afectada por varias especies de micoplasmas, pero Mycoplasma gallisepticum (MG) es el más significativo económicamente. La adherencia de los micoplasmas a las células epiteliales respiratorias del huésped, constituye un paso crítico en el camino que lleva a la infección y a la enfermedad, y se realiza mediante lipoproteínas localizadas en la superficie de la bacteria. En un estudio reciente in silico, se predijo la existencia de un grupo de posibles proteínas de superficie con potenciales propiedades antigénicas que podrían ser usadas como candidatas para explorar nuevas vacunas, así como pruebas diagnósticas. Uno de estos candidatos fue OppA. En este trabajo se describe el clonaje molecular de un segmento que codifica para un fragmento de OppA, un polipéptido de 233 aa y aproximadamente 30kDa, así como su expresión, purificación y la respuesta inmunogénica inducida por el producto obtenido.
Palabras clave: Mycoplasma gallisepticum, proteína de superficie, proteína recombinante, purificación por IMAC.
INTRODUCTION
Mycoplasmas, the simplest self-replicating organisms known, are distinguished phenotypically from other bacteria by their minute size and total lack of cell wall, characteristics derived from the degenerative (reductive) evolution from their gram-positive bacterial ancestors (1).
Despite their genetic simplicity, mycoplasmas are considered to be major animal and plant pathogens worldwide. The poultry industry is affected by several species of mycoplasmas. Of these, Mycoplasma gallisepticum (MG) is the most economically significant. The chronic respiratory disease caused by this infection represents serious problems. Although this disease does not necessarily account for high mortality, morbidity is high, and the consequent damaging effects of the disease are due to lowered egg production, retarded growth, poor carcass quality and predisposition to secondary viral and bacterial infections (2). Valuable breeding flocks that become infected may well be slaughtered or may lose the export market for their progeny (3).
The attachment of mycoplasmas to host respiratory epithelial cells constitutes a critical step in the pathway leading to infection and disease (4), and is achieved by lipoproteins localized on their surface. Due to the lack of cell wall and surface appendages common to other eubacteria, the mycoplasma membrane proteins are directly involved in mycoplasma-host interaction and play a crucial role in mycoplasma pathogenesis. The critical role of citadherence in virulence is reflected by the inability of noncytadhering MG strains to cause disease in experimentally infected animals (5). Although the adhesins play the major role in cytadhesion, the process also involves accessory membrane proteins. Data emerging from recent studies indicate that, although some putative cytadhesins like VlhA family (6), GapA (7), PvpA (8) or cytadhesin-related molecules like CrmA (9) from MG have been reported, several of them have yet to be identified, defined and characterized, a step which is crucial to understand the exact contribution of each of these proteins in promoting and maintaining a successful infection in the avian host.
Recently, in a bioinformatic study carried out in our laboratory, a fragment of OppA protein (locus_tag="MGA_0237") that encodes a polypeptide of 233 aa of about 30 kDa, was predicted as MG putative surface polypeptide with potential antigenic properties. Since they are in direct contact with the environment and, specifically, with the host immune system, surface localized virulence factors such as adhesins, serve as excellent immunogenic candidates (10). In this regard, this protein could be used as a candidate for exploring new vaccines and diagnostic systems.
In this work, we describe the molecular cloning of the DNA segment that codifies for this fragment, as well as its expression, purification and immunogenic capacity of the product obtained.
MATERIALS AND METHODS
Vectors, bacterial strains and growth conditions
The Top 10 F' and BL21-CodonPlus(DE3)-RIL cells were obtained from the strain bank of the Centre for Genetic Engineering and Biotechnology (CIGB) (La Habana, Cuba). The MG strain R was donated by Dr. Konrad Sachse (Friedrich-Loeffler-Institut, Fed. Res. Ctr. for Animal Health, Jena, Germany). Top 10 F' competent cells were transformed with pQE30 plasmid and grown O/N at 37°C in Luria-Bertani (LB) medium supplemented with 100mg of ampicillin/ml (Promega).
Vector Preparation
The pQE30 plasmid (QIAGEN) was purified using the AccuPrepTM Plasmid Extraction Kit, spin column (Hylabs). Five micrograms of the vector were digested with 50U each of Sac I and Kpn I (all the reagents from Promega) in 100ml of total volume and in presence of 0.1mg/ml BSA and 10ml of MULTI-CORETM buffer; the digestion was carried out for 3 hours at 37°C. The digested product was applied on a 0.8% (w/v) low- melting point agarose gel, from where, after electrophoresis, a 3455bp band corresponding to the digested pQE30 plasmid was isolated. This band was purified using the AccuPrep® Gel Purification Kit (Hylabs).
When kits were used all the procedures were carried out according to the manufacturer's instructions.
Obtainment and preparation of PCR products
The gene fragment was obtained by PCR using MG strain R chromosomal DNA as template and Pwo polymerase (CIGB) in a total volume of 50µl reaction. All the PCR conditions were adjusted according to the primer melting temperatures and PCR fragment length. The primers were designed in such a way that Sac I and Kpn I restriction sites were additionally introduced in the extremes 5´and 3´ of the PCR product for its subsequent, in frame, cloning in the expression vector pQE30. The final PCR product was purified by AccuPrep® PCR Purification Kit, spin column (Hylabs).
Five micrograms of the PCR product were digested with 50U each of Sac I and Kpn I (Promega) in 100ml of total volume and in presence of 0.1mg/ml BSA and 10ml of MULTI-CORETM buffer; the digestion was carried out for 3 hours at 37°C. The digested product was purified by AccuPrep® PCR Purification Kit, spin column (Hylabs).
Recombinant plasmid generation
To obtain the recombinant construction, the previously digested gene fragment and pQE30 (Qiagen) vector were ligated; 4U of T4 DNA Ligase (Promega), 2ml T4 DNA Ligase buffer 10x (Promega) were used in a total 20ml volume and the reaction was carried out at 22°C for 5h.
For transformation, Ca2+ competent cells from Top 10 F' of E. coli (CIGB) were used. They were transformed as follows: 10ml of the ligation product were added to 150ml of competent cells, the mix was incubated in ice for 20 min, then, 45 sec at 42°C and, to finish, again in ice for 5 min; 1ml of LB medium was added to the mix and this was incubated for 1h at 37°C at 50rpm. LB plates supplemented with 100mg of ampicillin/ml were inoculated with this culture and incubated O/N at 37°C.
The screening of recombinant clones was done by PCR with the same primers used for obtaining the fragment.
BL21-CodonPlus(DE3)-RIL competent cells (CIGB) were transformed with the recombinant plasmid following the same protocol used for transforming the Ca2+ competent cells from Top 10 F'.
Expression of recombinant fragments
For expression experiments, the method of autoinduction developed by Studier (13) with some modifications was used. E. coli BL21-CodonPlus (DE3)-RIL cells (CIGB) harboring the expression constructs were cultured O/N at 37°C in a Luria-Bertani broth supplemented with 50mg of kanamycin/ml. Next day, 100ml (in a 1l erlenmeyer) of auto-induction media (1% Tryptone; 0.5% Yeast extract; 25mM Na2HPO4; 25mM KH2PO4; 50mM NH4Cl; 5mM Na2SO4; 0.5% glycerol; 0.05% glucose; 0.2% alfa-lactose monohydrate; 2mM MgSO4 heptahydrate; 0.05mM FeCl3) supplemented with 100mg of ampicillin/ml (Sigma) were inoculated with 10ml of the O/N inoculum and grown for 16 hours with shaking of 310rpm. The cells were harvested by centrifugation at 5,000 x g for 10 min and conserved at -20°C.
Recombinant protein isolation
Cell pellets were resuspended in PBS and lysis carried out in a French press. The lysates were centrifuged at 5,000 x g for 10 min and the pellet treated with 8M urea for 3 hours at room temperature for protein solubilisation. The isolated soluble proteins were obtained by centrifugation at 10,000 x g for 30 min.
Polyclonal antibody production in rabbits
Two rabbits were immunized according to the following schema: immunization with 200mg subcutaneous in Complete Freund's Adjuvant (CFA) (Sigma); day 14, 100mg intradermal in Incomplete Freund's Adjuvant (IFA) (Sigma); day 28, 100mg subcutaneous with IFA; day 56, 50mg subcutaneous in IFA.
Rabbit polyclonal sera evaluation against recombinant proteins
Both rabbit polyclonal preimmune and postimmune sera, were evaluated by indirect ELISA; 1 µg of recombinant protein was immobilized in plates (MaxiSorp, NUNC) for 3h at 37ºC. Plates were blocked with BSA (SIGMA) 1% for 1h at 37ºC. Antibodies were diluted 1:1000, antirabbit-HRP (Sigma) conjugate 1:5000 and both assayed for 1h at 37ºC.
SDS-PAGE and Western blot
Briefly, SDS-PAGE was performed at constant current of 30mA on a 15% acrylamide gels. The proteins were analyzed either through staining with Coomassie Blue or by using Western blotting. For Western blots, the proteins were transferred to nitrocellulose membranes using a semi-dry transfer equipment. The membranes were blocked with 1% PBS-BSA for 1 hour. The blots were probed for 2 hours at 37°C, with rabbit recombinant protein antisera from experimentally infected rabbits. Bound antibodies were detected by using horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibodies (Sigma) and 4-chloro-1- naphthol-H2O2 substrate Kit (Bio-Rad), according to the manufacturer's instructions.
RESULTS AND DISCUSSION
The goal of this study was to obtain a recombinant fragment of the OppA, previously predicted in our laboratory, as a putative surface protein and to evaluate its immunogenic properties. Interestingly, an homologous of this protein in Mycoplasma hominis, has shown to play an important role in the pathogenesis of this bacterium (11, 12).
The 859 bp band corresponding to the oppA gene fragment was successfully obtained from MG strain R chromosomal DNA by PCR (Figure 1).
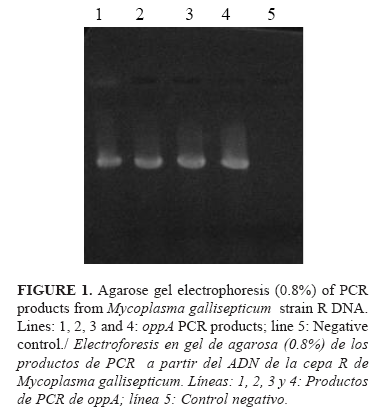
The oppA gene fragment was isolated from the PCR mixture and cloned into pQE30 vector using the Kpn I and Sac I restriction sites introduced into the amplified gene. One positive recombinant clone was detected by PCR analysis (Figure 2).The initial transformation was done in Top10-F' cells of E. coli because this strain facilitated the propagation and subsequent purification of plasmids, but for the expression, it was necessary to transform BL21(DE3) cells of E. coli with the recombinant plasmids in order to guarantee a greater control of the gene expression and a high level of it. In pQE plasmids, the gene expression is under the control of the T7 RNA polymerase. This enzyme is so specific, active, and processive that the amount of target RNA produced can be comparable to the amount of ribosomal RNA in a cell. A problem in using inducible T7 expression systems is that T7 RNA polymerase is so active that a small basal level can lead to a substantial expression of the target protein even in the absence of an inducer.
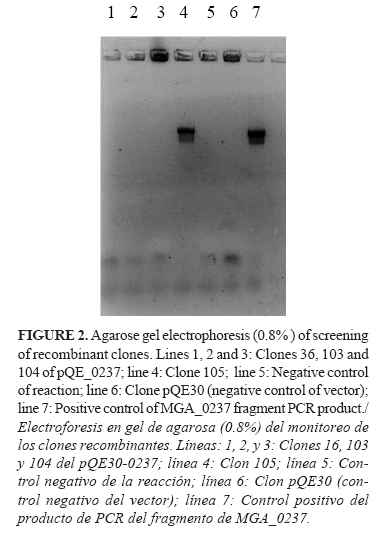
If the target protein is sufficiently toxic to the host cell, establishment of the target plasmid in the expression host may be difficult or impossible, or the expression strain may be unstable or accumulate mutations. In the BL21(DE3)-pQE system this problem is overcome, the lac operator sequence (the binding site for lac repressor) is placed in both, the start site of a T7 promoter (of the pET plasmid) and in the lac promoter, under the control of which is the T7 RNA polymerase in the genome of cells. In this way, the basal level of the target protein in uninduced cells is substantially reduced, but induction leads to the typical high levels of expression.
The obtained BL21(DE3) clones from the original recombinant plasmids were thus designated pQE30-oppA. This clone was then used for the expression of the recombinant fragment of the OppA protein by using the auto-induction method described by Studier (13). Auto-induction allows efficient screening of many clones in parallel for expression and solubility as cultures have only to be inoculated and grown to saturation, and yields of target protein are typically several folds higher than those obtained by conventional IPTG induction. In addition, IPTG, a very expensive reagent, is substituted by the cheaper a-lactose. In this system, lactose is used as inductor, and the unintended level of expression of the target protein in the initial moments of the culture is abolish by the addition of glucose to the media. Glucose prevents induction by lactose by well-studied mechanisms. In the original protocol, expression strains are grown O/N to saturation in non-inducing media, which is then inoculated into the autoinduction media (13). We substituted this non-inducing media by the normal LB because in our hands the yields were similar and the LB media was easier to prepare (14).
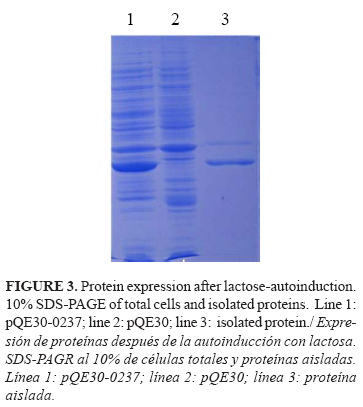
The SDS-PAGE analysis showed the expression of a 30kDa protein fragment. The same expression level was obtained for all the clones tested. From this point, we decided to continue working only with one of the clones, and for further analyses we named it pQE30-0237.
The expression of the OppA fragment for further purification steps was also done by the autoinduction method. The isolation of the OppA protein fragment was a simple process, facilitated by the expression in an insoluble form. The pellets were dissolved in 8M urea and, in these conditions, the solubilisation of the protein was achieved and it was obtained with a high level of purity (Fig. 3).
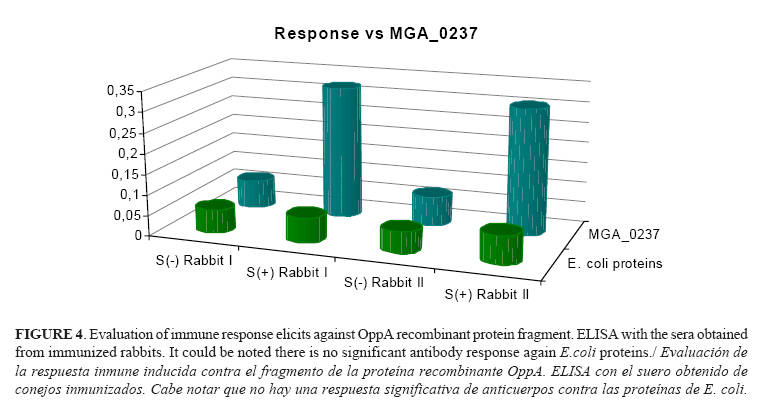
For the evaluation of the immunogenic properties of the OppA fragment protein, two female rabbits were immunized. Both animals developed specific immune response against OppA fragment protein (Fig. 4) as shown by the ELISA analysis. No antibodies were detected in the sera of the animal before immunizations [S(-)] and either was detected any recognition signal of E. coli proteins by sera of immunized rabbits [S(+)].
CONCLUSIONS
As conclusions of this work, we can say that the in silico predicted MG polypeptide OppA is immunogenic in rabbits.
The described methodology will permit us to count with large quantities of OppA antigen fragment, which could facilitate the further studies of the subcellular location of this protein and evaluate its role on MG pathogenesis mechanisms.
The potential surface localization of this protein, its already proved immunogenicity, and the involvement of its homolog from Mycoplasma hominis in virulence mechanisms of this mycoplasma, make it a good candidate to be tested in studies for vaccine development or diagnostic systems.
REFERENCES
1. Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev. 1998;62(4):1094-1156.
2. Kleven SH. Mycoplasmas in the etiology of multifactorial respiratory disease. Poult Sci. 1998;77(8):1146-1149.
3. Bradbury JM. Gordon Memorial Lecture. Poultry mycoplasmas: sophisticated pathogens in simple guise. Br Poult Sci. 2005;46(2):125-136.
4. Levisohn S. Early stages in the interaction between Mycoplasma gallisepticum and the chick trachea, as related to pathogenicity and immunogenicity. Isr J Med Sci. 1984;20(10):982-984.
5. Papazisi L, Frasca S, Gladd M, Liao X, Yogev D, Geary SJ. GapA and CrmA coexpression is essential for Mycoplasma gallisepticum cytadherence and virulence. Infect Immun. 2002;70(12):6839-6845.
6. Markham PF, Glew MD, Sykes JE, Bowden TR, Pollocks TD, Browning GF, et al. The organisation of the multigene family which encodes the major cell surface protein, pMGA, of Mycoplasma gallisepticum. FEBS Lett. 1994;352(3):347-352.
7. Goh MS, Gorton TS, Forsyth MH, Troy KE, Geary SJ. Molecular and biochemical analysis of a 105 kDa Mycoplasma gallisepticum cytadhesin (GapA). Microbiol. 1998;144(Pt 11):2971-2978.
8. Boguslavsky S, Menaker D, Lysnyansky I, Liu T, Levisohn S, Rosengarten R, et al. Molecular characterization of the Mycoplasma gallisepticum pvpA gene which encodes a putative variable cytadhesin protein. Infect Immun. 2000;68(7):3956-3964.
9. Papazisi L, Troy KE, Gorton TS, Liao X, Geary SJ. Analysis of cytadherence-deficient, GapA-negative Mycoplasma gallisepticum strain R. Infect Immun. 2000;68(12):6643-6649.
10.Chaudhuri R, Ramachandran S. Prediction of virulence factors using bioinformatics approaches. Methods Mol Biol. 2014;1184:389-400.
11.Hopfe M, Henrich B. OppA, the ecto-ATPase of Mycoplasma hominis induces ATP release and cell death in HeLa cells. BMC Microbiol. 2008;8:55.
12.Hopfe M, Dahlmanns T, Henrich B. In Mycoplasma hominis the OppA-mediated cytoadhesion depends on its ATPase activity. BMC Microbiol. 2011;11(1):185.
13.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207-34.
14.Agüero JA, Sánchez O, Barrera M, Toledo JR. Molecular cloning and expression of a fragment of the gene codifying for the protein Erns of the Classical Swine Fever Virus. Rev Salud Anim. 2008;30(2):85-92.
Recibido: 21-1-2015.
Aceptado: 5-3-2015.
*Corresponding author: José Antonio Agüero. E-mail: jaaguero@censa.edu.cu.


{kind=link}