










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Mycoplasma gallisepticum (M. gallisepticum) es el agente etiológico fundamental de la micoplasmosis aviar, enfermedad de curso crónico que afecta fundamentalmente a pavos y gallinas, aunque puede estar presente en varias especies de aves (patos, gansos, faisanes, codornices, perdices, gallinas de Guinea y palomas) (1). M. gallisepticum se considera el micoplasma más significativo desde el punto de vista económico para la avicultura, por las grandes pérdidas que ocasiona. Solo en los Estados Unidos las pérdidas han alcanzado los 150 millones de dólares anuales, mientras que a nivel global estas han estado alrededor de los 780 millones de dólares en igual periodo de tiempo (2).
La vacunación con bacterinas, o vacunas vivas, puede ser una opción en aquellos casos en los cuales el control de la infección no es factible con adecuadas estrategias de manejo. Aunque las vacunas proporcionan alguna protección contra la enfermedad y disminuyen las pérdidas económicas de los productores, aún ocurren brotes de la enfermedad, con frecuencias demasiado altas (3).
El esclarecimiento de los mecanismos patogénicos, por los cuales M. gallisepticum ejerce sus efectos sobre las aves de corral, es crítico en la búsqueda racional de mejores vacunas y estrategias de control. La adhesión de los micoplasmas al epitelio mucosal del tracto respiratorio de las aves es un requisito previo e indispensable para la infección y posterior desarrollo de la enfermedad, y se logra mediante lipoproteínas localizadas en su superficie (4,5). El papel crítico de la citadherencia en la virulencia se refleja en la incapacidad de las cepas de M. gallisepticum no adherentes para provocar la enfermedad en animales infectados experimentalmente (6). Esta capacidad está mediada por dos de las principales citadhesinas de M. gallisepticum, GapA y CrmA. Estudios recientes indican que, si bien se han descrito algunas otras citadhesinas como la familia VlhA (7) la proteína PvpA (8), existen muchos componentes de este tipo que todavía no han sido identificados, definidos y caracterizados (9,10). La identificación y la obtención de estas proteínas es de gran importancia para comprender los mecanismos patogénicos de los micoplasmas y la exacta contribución de cada una de ellas en la generación y el mantenimiento de una infección exitosa en el hospedero aviar; además, es crucial para el desarrollo de vacunas y de medios de diagnóstico.
En este trabajo se llevó a cabo el clonaje molecular de los genes que codifican para GapA y CrmA, así como su expresión y purificación, además de la evaluación de la antigenicidad de los productos obtenidos.
MATERIALES Y MÉTODOS
Vectores, cepas bacterianas y condiciones de cultivo de M. gallisepticum
La cepa DH5α de Escherichia coli (E. coli) (F- endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoRnupG purB20 φ80dlacZΔM15 Δ(lacZYA-argF) U169, hsdR17 (rK-mK+), λ-), se obtuvo del banco de cepas del Centro Nacional de Sanidad Agropecuaria (CENSA). La cepa ISM612 de E. coli fue suministrada por el Departamento de Microbiología Veterinaria y Medicina Preventiva, Colegio de Medicina Veterinaria, Universidad Estatal de Iowa, Ames, EUA. La cepa R(low) de M. gallisepticum fue donada por el Instituto Friedrich-Loeffler, Centro Federal de Investigaciones para la Salud Animal, Jena, Alemania. Para el clonaje molecular y la posterior expresión de los genes clonados se empleó el vector de fusión con poli-His, pQE30 (Qiagen).
La cepa R(low) de M. gallisepticum se cultivó en medio Hayflick caldo. El cultivo se incubó estáticamente a 37ºC, en presencia de CO2 al 5 % hasta que alcanzó una coloración amarilla. Posteriormente, se subcultivó en medio Hayflick agar donde se incubó durante siete días a 37ºC, en presencia de CO2 al 5 %. Se seleccionó una colonia aislada y se sembró nuevamente en medio Hayflick caldo. El cultivo se incubó estáticamente a 37ºC, en presencia de CO2 al 5 % hasta que alcanzó una coloración amarilla.
Diseño de los cebadores para el clonaje posterior de los genes
Los cebadores necesarios para obtener los fragmentos a clonar, mediante el empleo de la Reacción en Cadena de la Polimerasa (PCR, del inglés Polymerase Chain Reaction) se diseñaron con el empleo del programa Vector NTI Advance v 11.0 (Invitrogen). Como blanco se empleó la secuencia genómica de la cepa M. gallisepticumR (low), que se descargó de la base de datos del NCBI (Nº Acceso: AE015450.2, GI: 284811830).
En el diseño de los cebadores, sentido y antisentido, se incorporaron las secuencias correspondientes a los sitios de restricción de las enzimas SphI y SmaI, respectivamente, presentes en el sitio de clonaje múltiple del plásmido pQE30 y ausentes en las secuencias de los genes a clonar. A continuación, se relacionan las secuencias de los cebadores a emplear:
crmAF-ACATGCATGCAAGCAATCAGATAAGAGTAAC
crmAR-TCCCCCGGGTGTAGGCACTACTCATGAAG
gapAF-ACATGCATGCTCTGGAGCTAAGCCAAATAA
gapAR-TCCCCCGGGATAACCTGGAAGCGCATCAA)
En el diseño también se tuvo en cuenta eliminar del producto de amplificación, la región correspondiente a los péptidos-señal identificados con el empleo del SignalP y el LipoP.
Para determinar si con estos cebadores todos los productos de amplificación quedaban en el marco de lectura correcto en el plásmido receptor, se realizó in silico el clonaje de cada uno de los productos de PCR y se evaluó si se generaban las regiones codificantes esperadas. Todos los análisis de PCR, Clonaje y Expresión in silico se realizaron con el empleo del Vector NTI Advance v 11.0.
Extracción de ADN genómico
El ADN genómico se purificó según el protocolo empleado por Kang et al. (11), con algunas modificaciones. Se centrifugaron 5 mL de un cultivo de la cepa R(low) de M. gallisepticum, por 15 min a 3 000xg. La masa celular se resuspendió en 2,5 mL de tampón de lisis. La suspensión resultante se incubó 10 min a 50ºC, seguidos de 10 min a 37ºC. Se adicionaron 25 µL de proteinasa K (100 mg/mL) y se incubó por 40 min a 37ºC. A esta mezcla se le añadieron 5 µL de KAc 5M y se centrifugó 15 min a 14 000xg. El sobrenadante se transfirió a un nuevo tubo y se adicionaron 1775 µL de isopropanol; la mezcla se centrifugó a 14 000xg por 30 min. El precipitado se lavó con etanol al 70 % y se centrifugó 15 min a 14 000xg. El precipitado se dejó secar media hora a temperatura ambiente y se resuspendió de manera pasiva en 500 µL de H2Odd, por 60 min a 4ºC.
Los resultados de la purificación se monitorearon por electroforesis en gel de agarosa al 1 % (peso/volumen, p/v). La misma se llevó a cabo a 100 V (voltaje constante). Como agente de tinción se empleó una solución de bromuro de etidio (Sigma) (0,5 µg/mL).
Amplificación por PCR de los genes identificados
Los genes, o fragmentos de genes a clonar, se obtuvieron por amplificación mediante la PCR (12), con el empleo del ADN genómico de la cepa R(low) de M. gallisepticum, como molde de la reacción. Para la amplificación de los genes por PCR se adicionó 1 µL del ADN genómico de la cepa R(low) a la mezcla que contenía 5 µL de tampón de ADN polimerasa 10x (BioLabs), dNTPs (BioLabs) 200 µM (concentración final), ambos cebadores 400 nM (concentración final) y 0,2 U de la VentADN polimerasa (BioLabs) en un volumen total de 50 µL. El programa de PCR fue el siguiente: 1 ciclo de 94ºC por 5 min, seguido por 30 ciclos de 94ºC por 1 min, 45ºC por 1 min y 72ºC por 3 min y, por último, un ciclo de 72ºC por 7 min.
Los resultados de la PCR se monitorearon por electroforesis en gel de agarosa. En todos los casos se aplicaron 3 µL de patrón de peso molecular (1kb leader, Fermentans).
Generación de las construcciones recombinantes
Los genes se clonaron en el vector pQE30 de fusión con poli-His.
Preparación de los productos de PCR
Los productos de PCR se purificaron con el juego comercial “QIAquick PCR Purification Kit” (Qiagen), según protocolo del fabricante. La cuantificación de los productos de PCR purificados se realizó en un equipo GeneQuant II (Pharmacia-Biotech).
Se digirieron 4 µg de los productos de PCR con 10 U de SphI (10U/µL), en un volumen total de 20 µL, en presencia de 2 µL de tampón Tango 10x (Fermentans), a 37ºC durante 2 h. Pasado ese tiempo se añadieron 10 U de SmaI (10 U/µL) (Fermentans), y la mezcla se incubó por 2 horas más a 30ºC. Las enzimas se inactivaron incubando la mezcla 20 min a 65ºC.
Los productos de la digestión se purificaron con el empleo del juego comercial “QIAquick PCR Purification Kit” (Qiagen), siguiendo el protocolo del fabricante. La cuantificación final de los productos de PCR purificados se realizó en un equipo GeneQuant II (Pharmacia Biotech).
Los resultados se monitorearon por electroforesis en gel de agarosa. En todos los casos se aplicaron 3 µL de patrón de peso molecular (1kb leader, Fermentans).
Preparación del vector
Se inocularon 5mL de medio Luria Bertani (LB), suplementado con ampicillina a una concentración de 100 µg/mL, con una colonia de la cepa DH5α de E.coli contentiva del plásmido pQE30. El cultivo se incubó en agitación toda la noche a 37ºC. Con 4 mL de este cultivo, se inocularon 200 mL de medio LB (100 µg/mL de ampicilina). Este cultivo también se incubó en agitación toda la noche a 37ºC. El vector se purificó con el juego comercial Plasmid Midi Kit (QIAGEN), según el protocolo descrito por el fabricante. El plásmido resultante, obtenido de los 200 mL de cultivo, se resuspendió en un volumen final de 300 µL de H 2 Odd.
Se digirieron 100 µg del vector. La digestión se realizó con 100 U de Sph I (10 U/µL) (Fermentans), en un volumen total de 300 µL, en presencia de 30 µL de tampón Tango 10x (Fermentans), a 37ºC durante 2 h. Pasado ese tiempo, se añadieron 100 U de Sma I (10 U/µL) (Fermentans); la mezcla se incubó por 2 horas más a 37ºC. Las enzimas se inactivaron incubando la mezcla 20 min a 65ºC. Los productos de digestión se purificaron con el empleo del juego comercial QIAquick Gel Extraction Kit (QIAGEN), según protocolo del fabricante.
Los resultados se monitorearon por electroforesis en gel de agarosa. En todos los casos se aplicaron 3 µL de patrón de peso molecular (1kb leader, Fermentans). La cuantificación del ADN plasmídico se realizó en un equipo GeneQuant II (Pharmacia-Biotech), luego de verificar por electroforesis la ausencia total de ARN en las muestras.
Ligazón de los genes con el vector
Para obtener las construcciones, los productos de PCR digeridos se ligaron (13) con 50 ng del vector pQE30 digerido, en una relación de tres moles de inserto por cada mol de vector. En cada caso se emplearon 400 U de T4 ADN ligasa (400 U/µL) (BioLabs) y 2 µL de tampón T4 ADN ligasa 10x (BioLabs), en un volumen total de 20 µL y la ligazón se realizó a 22ºC durante toda la noche. La cantidad de inserto en cada reacción se calculó según la fórmula:
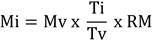
Transformación química de E. coli
Células quimio-competentes de la cepa DH5α de E. coli se transformaron (14) con el producto de la ligazón de los genes y el vector pQE30. Se empleó la cepa DH5α de E. coli debido a que esta facilita la propagación y posterior purificación de los plásmidos.
Para la transformación se descongelaron las células competentes en hielo por 10-15 min. A 100 µL de células competentes se le adicionaron 10 µL del producto de la ligazón y se mezclaron suavemente. Como control se empleó el plásmido pQE30, del cual se adicionaron 20 ng, también a 100 µL de células competentes. Se incubó en hielo durante 20 min; para incrementar la eficiencia de la transformación se dio un choque térmico a 42°C durante 45seg, seguido de una incubación en hielo de 5 min. A cada alícuota se le adicionó 1 mL de LB y se incubó 1 h a 37°C con agitación a 50 rpm. En placas de LB suplementadas con ampicilina (Sigma) (100 µg/mL) se sembraron 150 µL de los cultivos de la cepa transformada con los diferentes productos de ligazón y en otra se sembraron 50 µL del cultivo del plásmido control. Las placas se incubaron toda la noche a 37ºC.
Monitoreo de las construcciones recombinantes
En un primer paso de filtrado se seleccionaron los plásmidos con una talla superior a la talla del pQE30 (potenciales clones recombinantes). Para esto los posibles plásmidos recombinantes se purificaron por el método de lisis alcalina, según lo descrito por Maniatis et al. (15), a partir de cultivos en 3 ml de caldo LB suplementado con ampicillina (100 µg/mL). El precipitado final se resuspendió en 30 µL de H2Odd.
La presencia de los insertos en los plásmidos de mayor talla que el pQE30, se confirmó por PCR según el protocolo descrito previamente. Para los PCR se tomaron 2 µL de cada uno de los plásmidos purificados; se empleó la enzima Taq-Pol (Promega) a una concentración de 1U/reacción y se utilizaron los mismos cebadores que para la amplificación inicial de los genes a partir del ADN genómico de M. gallisepticum.
Los resultados se monitorearon por electroforesis en gel de agarosa. En todos los casos se aplicaron 3 µL de patrón de peso molecular (1 kb leader, Fermentans).
Obtención de los polipéptidos recombinantes a partir de las construcciones generadas
Para la expresión de los genes clonados se empleó la cepa ISM612 de E. coli.
Transformación de las cepas de expresión
Tanto para la preparación de las células competentes como para la transformación de la cepa ISM612 de E.coli, se siguieron los protocolos descritos previamente. Las placas con LB sólido se suplementaron adicionalmente con cloranfenicol (Sigma) (35 µg/mL).
La presencia de las construcciones recombinantes en las células transformadas se monitoreó por PCR. Se sembraron las colonias en 1mL de caldo SB suplementado con ampicillina (100 µg/mL) y cloranfenicol (35 µg/mL); se incubaron en agitación toda la noche a 37ºC. Se tomaron 100 µL de los cultivos, se centrifugaron 5 min a 1 000 xg y se reconstituyeron en 100 µL de H2Odd. La suspensión se calentó 10 min a 95ºC y se volvió a centrifugar 5 min a 10 000 xg. El PCR se llevó a cabo según el protocolo descrito previamente, tomando 5 µL de cada uno de los sobrenadantes. Se empleó la enzima Taq-Pol (Promega) a una concentración de 1 U/µL. Se utilizaron los mismos cebadores que para la amplificación inicial de los genes a partir del ADN genómico de M. gallisepticum.
Los resultados se monitorearon por electroforesis en gel de agarosa. En todos los casos se aplicaron 3 µL de patrón de peso molecular (1kb leader, Fermentans).
Expresión de los genes recombinantes
La expresión se realizó mediante inducción con isopropil-β-D-tiogalactopiranosido (IPTG) (Sigma). Se prepararon inóculos de las construcciones recombinantes en la cepa ISM612, en 10 mL de caldo SB suplementado con ampicillina (Sigma) (100 µg/mL) y cloranfenicol (Sigma) (35 µg/mL); estos se incubaron en agitación toda la noche a 37ºC. Los inóculos se añadieron a 500 mL de caldo SB y se incubaron en agitación a 37ºC. Cuando estos alcanzaron una DO a 600 nm de 0,4÷0,6, se indujo la expresión con IPTG 1 mM. La inducción se prolongó por cuatro horas en agitación a 37ºC.
La evaluación del resultado de la inducción de la expresión se realizó por Western-Blott. Las muestras se centrifugaron 10 min a 6000 xg, de 1 mL de los cultivos; luego la masa celular se resuspendió en 100 µL de H2Odd. Finalmente, de estos se tomaron 10 µL a los que se le adicionaron 2 µL de tampón de muestra 6x (Promega); todas las muestras se hirvieron durante 10 min antes de aplicar en el gel.
Purificación de los polipéptidos recombinantes de M. gallisepticum
Extracción y solubilización de los productos recombinantes
La masa celular obtenida después de la inducción de cada cultivo de 500 mL se resuspendió en 75 mL de PBS. Las células se lisaron en un equipo Vibracell (Sonics&Materials), al someterlas a seis ciclos de sonicación de 10 seg con intervalos de 10 seg entre cada ciclo (60 kHz, corriente constante). El producto de la lisis se centrifugó 30 min a 14 000 xg.
El sobrenadante de cada muestra se desechó y los restos celulares sedimentados se resuspendieron en 75 mL de PBS-SDS-1 %. La mezcla se incubó, en agitación moderada, por una hora a temperatura ambiente. Posteriormente, se centrifugó por 30 min a 14 000 xg. El sedimento se desechó y el sobrenadante se incubó por 30 min a 4ºC. La mezcla se centrifugó por 30 min a 14 000xg. Los restos celulares sedimentados se resuspendieron en 75 mL de PBS-SDS-1 %.
La presencia de las proteínas de interés en la fracción soluble se monitoreó por SDS-PAGE y Western-Blott. Se tomaron 10 µL de las muestras (tanto de las fracciones insolubles como de las solubles) a los que se les adicionaron 2 µL de tampón de muestra 6x (Promega); todas las muestras se hirvieron durante 10min antes de aplicar en el gel.
Purificación por Cromatografía de Aninidad por Metal Inmovilizado (IMAC, del inglés Inmobilized Metal Affinity Chromatography)
Para purificar los productos recombinantes por cromatografía de afinidad por metales inmovilizados (IMAC), se cargaron las columnas con 1 mL de Chelating Sepharose Fast Flow (Pharmacia-Biotech) y la purificación se llevó a cabo según instrucciones del fabricante. Las columnas se equilibraron con cinco volúmenes (5 mL) de tampón de equilibrio. Se aplicaron 20 mL de cada uno de los extractos de proteínas por columna. Se lavaron con 10 volúmenes de tampón de lavado. Las proteínas se eluyeron con 5 mL de tampón de elusión (se colectaron fracciones de 1,5 mL). Todas las soluciones se pasaron por filtros de 0.45 µm. El proceso se desarrolló con un flujo determinado por la gravedad.
La evaluación de los resultados del proceso de purificación se realizó por SDS-PAGE y Western-Blott. Las muestras se prepararon tomando 10 µL de cada fracción, a los que se le adicionaron 2 µL de tampón de muestra 6x (Promega); todas las muestras se hirvieron durante 10 min antes de aplicar en el gel.
Monitoreo de la expresión, extracción y solubilizaciónpor SDS-PAGE y Western-Blott
La electroforesis se realizó bajo condiciones reductoras (16) y se corrió a corriente constante con 15 mA, en geles de acrilamida al 12 % (p/v). Los geles se tiñeron con solución de Coomasie durante 10 min en agitación lenta. Posteriormente, los geles se destiñeron hirviéndolos en H2Odd hasta observar las bandas de proteínas definidas.
Para el Western-Blott se hizo una transferencia semiseca (17) de las proteínas del gel de poliacrilamida a una membrana de nitrocelulosa. La transferencia se realizó a corriente constante y esta se calculó multiplicando el área del gel que se iba a transferir por 2,5. La membrana se bloqueó durante una hora en una solución de PBS-Tween 20 0,05 %-BSA 1 % y luego se incubó dos horas a 37ºC con un anticuerpo monoclonal contra poli-histidina (Sigma) diluido 1/2 000 en PBS-Tween 20 0,05%-BSA 1%. Seguidamente se incubó una hora a 37ºC con el conjugado enzimático anti IgG de ratón conjugada con fosfatasa alcalina (Sigma) diluido 1/5 000 en PBS-Tween 20 0,05 %-BSA 1 %, y por último la reacción se reveló con NBT-BCIP (Sigma) en tampón para fosfatasa alcalina.
Evaluación de la antigenicidad
Los ensayos se realizaron con las proteínas purificadas, el antisuero empleado fue un suero policlonal obtenido en conejos contra proteínas totales de la cepa R(low) de M. gallisepticum (donado por el Instituto Bacteriología y Micología de la Universidad de Medicina Veterinaria de Viena, Austria).
Evaluación de la antigenicidad en condiciones desnaturalizantes de los polipéptidos recombinantes purificados
La electroforesis y el Western-Blott se llevaron a cabo como se describió previamente. Para este último se empleó un anticuerpo policlonal obtenido en conejo, contra la cepa R(low) de M. gallisepticum, diluido 1/2 000 y un conjugado enzimático anti IgG de conejo conjugada con fosfatasa alcalina (Sigma) diluido 1/5 000.
Evaluación de la antigenicidad de los polipéptidos recombinantes purificados en condiciones nativas
Este experimento se realizó por ELISA para evaluar si las proteínas recombinantes eran capaces de reaccionar en condiciones nativas, con los anticuerpos contra la cepa R(low) de M. gallisepticum.
El protocolo que se siguió para el ELISA fue el siguiente. Las placas de ELISA (MaxiSorp, NUNC) se sensibilizaron con 100 µL de las proteínas de membrana diluidas en tampón de cubrimiento a una concentración de 1 µg/mL (cada muestra por duplicado), durante tres horas a 37ºC. Las placas se bloquearon durante una hora con 150 µL de una solución de PBS-Tween 20 0,05 %-BSA 1 % a 37ºC y luego se incubaron dos horas a 37ºC con 100 µL del suero policlonal contra la cepa R(low) de M. gallisepticum diluido 1/500, en PBS-Tween 20 0,05 %-BSA 1 %. Seguidamente se incubaron una hora a 37ºC con 100 µL de un conjugado enzimático anti IgG de conejo, conjugado con peroxidasa (Sigma) diluido 1/5 000 en PBS-Tween 20 0,05 %-BSA 1 %. Por último, la placa se reveló con 100 µL de tetra metil bencidina (TMB) (Sigma) en tampón sustrato peroxidasa a una concentración de 0,01 % y se detuvo la reacción con 50 µL de solución de parada antes de leerla a 450 nm. Se consideraron positivas aquellas muestras cuya DO fuera superior a dos veces la DO del blanco, constituido por un lisado de E. coli sometido al mismo proceso de purificación al que se sometieron los productos recombinantes.
RESULTADOS Y DISCUSIÓN
Amplificación por PCR de los genes identificados
Al emplear como molde el ADN genómico de la cepa R(low) de M. gallisepticum, se amplificaron los genes evaluados (Figura 1). En todos los casos se obtuvieron productos con las tallas esperadas (gapA 2926 y crmA 2734 pb). Es de significar que en el caso de gapA, además del producto esperado, se obtuvieron dos productos adicionales de 250 y 1400 pb. Este resultado se puede explicar porque se empleó un programa único, con una baja temperatura de unión que tenía como objetivo lograr la amplificación de todos los fragmentos, con independencia de la temperatura de fusión de los cebadores empleados. En los PCR, las bajas temperaturas de unión con respecto a las temperaturas de fusión de los cebadores pueden provocar inespecificidad en los ensayos: las primeras no deben estar por debajo de los 5ºC con respecto a las segundas (18). En este trabajo se empleó 45ºC como temperatura de unión, mientras la temperatura de fusión de los cebadores específicos, a partir de los cuales se generaron productos inespecíficos, se encontraban en el intervalo entre 60,8ºC y 81,9ºC. Tales diferencias pudieron haber provocado los resultados observados. Quizá están provocados, también, por fenómenos de recombinación que son muy frecuentes en los micoplasmas (19,20).
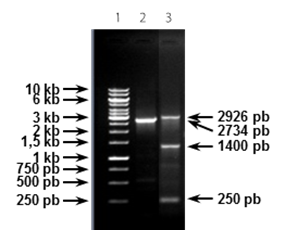
FIGURA 1 Obtención de los genes a clonar. Electroforesis en agarosa 1 % de los productos de PCR con los cebadores específicos para los genes gapA y crmA. Línea 1: MPM-1kb, Línea 2: crmA, Línea 3: gapA / Obtainment of the genes to be cloned. Electrophoresis in agarose 1 % of the PCR products with the specific primers for the gapA and crmA genes. Line 1: MPM-1kb, Line 2: crmA, Line 3: gapA
Como resultado de estas amplificaciones inespecíficas, se pueden obtener construcciones recombinantes con fragmentos de la secuencia esperada, o insertos cuyas secuencias no están relacionadas con los genes de interés. En el primer caso, el polipéptido recombinante podría conservar la utilidad de la proteína completa. Previamente existen ejemplos de clonaje de fragmentos de genes de micoplasmas para mapear su antigenicidad y sus dominios funcionales (21). Adicionalmente, el hecho de que los productos inespecíficos sean minoritarios con respecto al esperado, hace que las posibilidades de clonar uno de estos fragmentos sean inferiores a las de clonar las secuencias completas.
Generación de las construcciones recombinantes
La evaluación preliminar de la transformación inicial, que se realizó con propósitos de monitoreo del clonaje y para, posteriormente, contar con clones de propagación de los plásmidos recombinantes, se realizó mediante la comparación de la talla de las construcciones obtenidas con la del plásmido pQE30 nativo. Se identificaron un potencial clon recombinante para gapA y dos para crmA. El análisis de estas construcciones por PCR demostró que todas eran recombinantes (Figura 2).
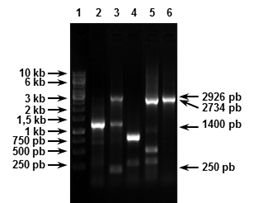
FIGURA 2 Monitoreo de los clones recombinantes. Electroforesis en agarosa 1 % de los PCR de las potenciales construcciones recombinantes. Línea 1: MPM-1kb, Línea 2: pQE30-gapA-17, Línea 3: gapA, Línea 4: pQE30-crmA-13, Línea 5: pQE30-crmA-16, Línea 6: crmA/ Monitoring of the recombinant clones. Electrophoresis in agarose 1 % of the PCR of the potential recombinant constructions. Line 1: MPM-1kb, Line 2: pQE30-gapA-17, Line 3: gapA, Line 4: pQE30-crmA-13, Line 5: pQE30-crmA-16, Line 6: crmA
Como se puede apreciar, solo una de las dos posibles construcciones para crmA poseía el inserto con la talla esperada, mientras el inserto de gapA tenía una talla menor (Figura 2, Tabla 1).
TABLA 1 Monitoreo de los posibles clones recombinantes. Comparación de la talla de los productos de PCR obtenidos con la talla esperada/ Monitoring of possible recombinant clones. Comparison of the size of the PCR products obtained with the expected size.
| Construcción | Talla esperada | Tallaobtenida |
|---|---|---|
| pQE30-crmA-13 | 2734 | 900 |
| pQE30-crmA-16 | 2734 | 2734 |
| pQE30-gapA-17 | 2926 | 1400 |
Este resultado es consecuencia directa de los múltiples productos que se obtuvieron en el paso de amplificación de los genes a partir del ADN cromosomal. La variedad de fragmentos se transfirió hasta la etapa de ligazón y clonaje y, probablemente, fue la causa del resultado obtenido. Las potenciales construcciones recombinantes, generadas sobre la base de estos errores, fueron filtradas más adelante, por el propio proceso al no generar proteínas recombinantes o no ser reconocidas por los anticuerpos contra M. gallisepticum. La secuenciación de los insertos podría aclarar la verdadera causa de lo observado.
Obtención de los polipéptidos recombinantes a partir de las construcciones generadas
En los experimentos de expresión se excluyó la construcción pQE30-crmA-13, ya que a partir de esta se generó un producto de PCR mucho más pequeño que lo esperado.

FIGURA 3 Expresión de las construcciones recombinantes. Western-Blot con anticuerpo monoclonal específico a péptidos de poli-histidina (antecedido por SDS-PAGE 12 %), de las células totales después de la inducción con IPTG de los cultivos de los clones recombinantes seleccionados. Línea 1: pQE30, Línea 2: pQE30-crmA-16, Línea 3: pQE30-gapA-17, Línea 4: PPM/ Expression of the recombinant constructions. Western Blot with monoclonal antibody specific to poly-histidine peptides (preceded by 12 % SDS-PAGE) of the total cells after induction with IPTG of the cultures of the selected recombinant clones. Line 1: pQE30, Line 2: pQE30-crmA-16, Line 3: pQE30-gapA-17, Line 4: PPM.
La expresión de las proteínas de micoplasmas en E. coli es una tarea complicada y difícil, debido al particular uso de codones de los mismos. Como consecuencia de los bajos niveles de expresión que se obtuvieron con el empleo de la cepa ISM612 de E. coli, fue imposible monitorear el proceso de expresión por SDS-PAGE y fue necesario emplear el Western-Blott con un anticuerpo monoclonal específico para péptidos de poli-histidina, debido a su mayor sensibilidad. Así se pudo detectar la presencia de productos recombinantes (Figura 3).
Como se puede apreciar, en ambos casos se obtuvo más de un fragmento. A pesar de que la cepa ISM612 de E. coli contiene un supresor opal inducible (trpT), así como la mutación prfB3 del factor 2 de liberación y que se ha empleado para expresar epítopos de la proteína P1 de M. pneumoniae (22), así como y para evaluar una librería genética de M. hyopneumoniae (23), con la cual se logró incrementar el reconocimiento de productos recombinantes de micoplasmas con anticuerpos (22), con esta cepa, no siempre se logra la expresión de los genes recombinantes, y cuando se consigue, generalmente es a bajos niveles. También es muy común la generación no solo de productos de la talla esperada, sino de tallas intermedias (22,23). Esto se explica porque la cepa ISM612 de E. coli no es capaz de impedir, en su totalidad, la terminación prematura de la traducción a causa de la presencia de los codones UGA. Por esta razón se obtienen proteínas truncadas con tallas que se corresponden con la aparición de estos codones opal. Incluso, en algunos casos, este mismo fenómeno puede explicar la no expresión de los genes.
Se han empleado otras variantes para expresar genes de micoplasmas, como la expresión en otros Mollicutes (24) y, en los casos en que pocos codones estén presentes en la secuencia a clonar, la mutagénesis sitio-dirigida (25); pero todas estas estrategias han brindado, de manera general, resultados poco satisfactorios y, en el caso particular de esta última, consume mucho tiempo y es cara, pues su costo crece con el número de codones UGA a mutar (26).
En el futuro se puede pensar en la obtención de los antígenos recombinantes mediante la generación de genes sintéticos (27,28). Este procedimiento, aunque es aparentemente más caro, posee dos ventajas significativas: simplifica extraordinariamente el trabajo y, por tanto, el tiempo necesario para obtener los resultados. Por otro lado, además de eliminar el problema de la presencia de los codones opal en la secuencia del gen a expresar, permite muy altos niveles de generación del producto recombinante, ya que en el proceso de preparación de estas construcciones, se optimizan los genes para su expresión en el sistema heterólogo seleccionado (29). Esto incrementa los rendimientos del proceso de obtención de los productos recombinantes. De esta manera podrían obtenerse los productos, que en esta oportunidad no fue posible expresar y, además, los rendimientos serían mucho más altos y la calidad de los productos recombinantes, superior.
Purificación de los polipéptidos recombinantes de M. gallisepticum
Extracción y solubilización de los productos recombinantes
Los altos niveles de expresión de las proteínas recombinantes usualmente conllevan la acumulación de la proteína en forma de agregados insolubles, denominados cuerpos de inclusión (CI). Debido a que, durante mucho tiempo, los CI se han considerado productos de desecho esencialmente formados por proteínas mal plegadas, propensas a agregarse, se han realizado importantes esfuerzos para eliminar, o al menos parcialmente disminuir su formación. Sin embargo, en los últimos años el escenario ha cambiado completamente. Recientemente se ha demostrado que las proteínas incluidas en CI no están necesariamente mal plegadas, sin actividad biológica y, por ende, pueden ser empleadas con fines biomédicos (30,31). No obstante, para cualquier uso terapéutico posterior es recomendable solubilizar y renaturalizar las proteínas in vitro.
Por los bajos niveles de expresión de los genes, podía suponerse la generación de los productos en forma soluble; como se puede apreciar en la Figura 4, esto no ocurrió así, probablemente porque se trataba de proteínas de membrana las cuales son particularmente insolubles. Por su alta hidrofobicidad fue necesario someter a pasos de solubilización a los productos generados.
Una de las ventajas de la obtención de las proteínas en CI es que la mayor parte de las proteínas del hospedero se expresa de forma soluble y una simple centrifugación, a continuación de la lisis celular, permite eliminar la mayor parte de los contaminantes sin la necesidad de emplear complicadas y costosas técnicas de purificación. La solubilización de las proteínas con SDS permitió la obtención de las proteínas puras de manera soluble, sin necesidad de la aplicación de complicados y poco eficientes procesos de renaturalización.
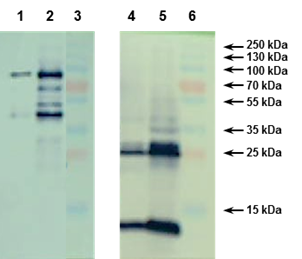
FIGURA 4 Solubilización de los productos recombinantes. Western-Blot con anticuerpo monoclonal específico a péptidos de poli-histidina (antecedido por SDS-PAGE 12 %) de las fracciones de solubilización. Línea 1: pQE30-gapA-17 (Se), Línea 2: pQE30-gapA-17 (S), Línea 3: PPM, Línea 4: pQE30-crmA-16 (Se), Línea 5: pQE30-crmA-16 (S), Línea 6: PPM. Donde Se-Fracción Sedimentada y S-Fracción Soluble/ Solubilization of recombinant products. Western Blot with monoclonal antibody specific to poly-histidine peptides (preceded by SDS-PAGE 12 %) of the solubilization fractions. Line 1: pQE30-gapA-17 (Se), Line 2: pQE30-gapA-17 (S), Line 3: PPM, Line 4: pQE30-crmA-16 (Se), Line 5: pQE30-crmA-16 ( S), Line 6: PPM. Where Se- Sedimented Fraction and S- Soluble Fraction.
A diferencia del resto de los agentes caotrópicos que se emplean en la solubilización de las proteínas recombinantes, el SDS interfiere en la purificación por IMAC (32). Este paso de eliminación de los agentes solubilizantes del sistema es particularmente complicado cuando se emplean agentes caotrópicos, como urea o hidrocloruro de guanidinio. Sin embargo, cuando se emplea SDS para solubilizar las proteínas, un protocolo descrito recientemente (32,33) soluciona este problema de una manera muy sencilla, la simple incubación de la muestra por 30 min a 4ºC y su posterior centrifugación antes de comenzar la purificación por IMAC. Ni las proteínas del hospedero ni las recombinantes coprecipitan con el SDS (32). Otras ventajas del empleo de este método consisten en la velocidad y la fácil ejecución del procedmiento y el bajo costo del SDS, con respecto al empleo y el costo de la urea o el hidrocloruro de guanidinio.
El hecho de obtener las proteínas bajo condiciones nativas es un importante resultado, ya que facilita los siguientes experimentos con los antígenos.
Purificación por IMAC
Aunque se realizó la ruptura celular antes del proceso de solubilización con SDS y disminuyeron considerablemente los niveles de impurezas de las muestras, sobre todo de las proteínas solubles citoplasmáticas, se incluyó un paso de purificación por IMAC para incrementar estos niveles, pero en lo fundamental, para aprovechar la propiedad que tienen estos sistemas de concentrar las muestras. Este aspecto era muy importante debido al bajo nivel de producción de las proteínas recombinantes en ISM612.
Los resultados de la purificación se observan en la Figura 5. Como se aprecia, las proteínas se eluyeron de las columnas, luego del sencillo y rápido proceso cromatográfico que solo incluyó tres soluciones tampón.

FIGURA 5 Purificación de los productos recombinantes. SDS PAGE 12%. Línea 1: pQE30-gapA-17, Línea 2: PPM, Línea 3: pQE30-crmA-16, Línea 4: PPM/ Purification of recombinant products. SDS PAGE 12%. Line 1: pQE30-gapA-17, Line 2: PPM, Line 3: pQE30-crmA-16, Line 4: PPM.
El proceso de purificación, en ausencia total del agente solubilizante (SDS), provocó la precipitación de los productos recombinantes, fenómeno que suele ocurrir cuando se trabaja con los agentes caotrópicos antes mencionados. Adicionalmente, permitió la concentración de los productos, aspecto este sobre todo importante cuando se emplea un sistema como la cepa ISM612, donde los niveles de expresión son bajos. Como se puede apreciar, a diferencia de lo que ocurría luego del paso de inducción de la síntesis de los productos recombinantes, después de la purificación GapA y CrmA ya son visibles por SDS-PAGE.
Evaluación de la antigenicidad de los polipéptidos recombinantes
Como se puede apreciar en la Figura 6, el resultado de este ensayo mostró que las dos proteínas evaluadas, GapA y CrmA, reaccionaron con el antisuero, lo que implica que ambas conservan sus propiedades antigénicas después de concluido todo el proceso de obtención y purificación, al menos a nivel de epítopos lineales, que son los únicos que se conservan en la técnica de Western-Blott.

FIGURA 6 Antigenicidad de los productos recombinantes. Western-Blot con anticuerpo policlonal específico a proteínas totales de la cepa R (low) de M. gallisepticum. Línea 1: pQE30, Línea 2: pQE30-gapA-17, Línea 3: pQE30-crmA-16, Línea 4: PPM/ Antigenicity of recombinant products. Western blot with polyclonal antibody specific to total proteins of the R (low) strain of M. gallisepticum. Line 1: pQE30, Line 2: pQE30-gapA-17, Line 3: pQE30-crmA-16, Line 4: PPM.
Para complementar este resultado y evaluar si las proteínas eran también reconocidas en condiciones nativas, se evaluó la reacción por ELISA. Los resultados confirmaron los obtenidos por Western-Blott. Tanto GapA como CrmA fueron reconocidas por el antisuero contra la cepa R(low) de M. gallisepticum y, adicionalmente, sugieren que los productos recombinantes conservan epítopos conformacionales, lo que es de gran importancia ya que, como ha sido descrito previamente, aproximadamente el 90 % de los epítopos B son de naturaleza conformacional (34).
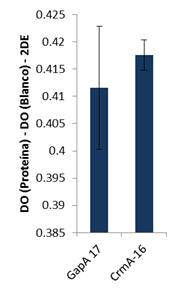
FIGURA 7 Antigenicidad de los productos recombinantes. ELISA con un Ac contra células completas de la cepa R(low) de M. gallisepticum. DO es la densidad óptica y DE es la desviación estándar del blanco/ Antigenicity of recombinant products. ELISA with an Ac against whole cells of the R (low) strain of M. gallisepticum. DO is the optical density and DE is the standard deviation of the target.
Las proteínas de superficie bacterianas son las que tienen las mayores posibilidades de interactuar con el sistema inmune y de estimular una respuesta efectiva (35); por esta razón se han desarrollado vacunas basadas, únicamente, en las proteínas de membrana (36) y son el blanco principal en los estudios de vacunología reversa (37).
La incorporación de GapA a la vacuna ts-11 incrementó la inmunogenicidad del preparado vacunal, así como los niveles de protección contra la enfermedad de los pollos vacunados (38,39). Por otro lado, se demostró que la coexpresión de esta y CrmA es vital para la virulencia del microorganismo (6). Este comportamiento las convierte en excelentes controles en los estudios de identificación de nuevos blancos para las investigaciones en el campo de los mecanismos de patogenicidad y del desarrollo de vacunas contra este microorganismo
En resumen, se logró la expresión de los genes que codifican para dos de las principales citadhesinas de M. gallisepticum, GapA y CrmA, aunque los productos se obtuvieron truncados, debido, en lo fundamental, a las dificultades de la expresión de genes de micoplasma en E. coli. En el futuro esta limitación se puede superar con el empleo de genes sintéticos, los cuales están optimizados para la expresión en el hospedero seleccionado (29). Así se garantizarían altos niveles de expresión de todos los genes, además de reducir significativamente el tiempo de investigación. El empleo de SDS como agente solubilizante facilitó la purificación de los productos recombinantes en condiciones nativas, reduciendo la complejidad del proceso e incrementando sus rendimientos, con respecto a los que se hubieran obtenido con el empleo de agentes como la urea. Contar con dos de los principales factores de virulencia de esta bacteria, obtenidos por vía recombinante, podría ser de gran utilidad en futuros estudios de los mecanismos de patogenicidad de la misma, ya que podrían servir como controles positivos en la búsqueda de nuevas de estas moléculas que permitirían realizar una análisis más certero y riguroso de los resultados. Además, se podría evaluar su papel en el desarrollo de sistemas serológicos de diagnóstico.

