










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.15 n.1 Ciudad de la Habana ene.-abr. 1999
Artículos de revisión
Instituto de Hematología e InmunologíaLeucemia linfoide crónica. Aspectos clínicos y biológicos
Dr. Porfirio Hernández RamírezResumen
La leucemia linfoide crónica (LLC) es el tipo de leucemia más frecuente en los individuos caucásicos. Su incidencia tiene una gran variación de acuerdo con el área geográfica. La variedad más frecuente es la de linfocitos B y es la que se describe en este trabajo. Su etiología es desconocida. En general la LLC puede presentar un amplio espectro de manifestaciones clínicas y diferentes complicaciones. Aproximadamente en el 10 al 30 % de los casos se produce una transformación en un proceso linfoproliferativo más agresivo. La LLC es una enfermedad heterogénea en su aspecto citomorfológico y puede además presentar variaciones en sus características inmunofenotípicas, citogenéticas y moleculares. Mediante las modernas técnicas moleculares se ha demostrado una mayor incidencia de trastornos citogenéticos que con los métodos tradicionales, los más frecuentes trisomía 12, deleciones 11q y del 13q12-14. Hasta el momento los estudios moleculares realizados no han podido identificar un oncogen asociado al desarrollo de la LLC; sin embargo, se mantiene un activo trabajo de investigación en este sentido.Descriptores DeCS: LEUCEMIA CRONICA DE CELULA-B.
Introducción
La leucemia linfoide crónica (LLC) es un síndrome linfoproliferativo crónico que se caracteriza por la acumulación de linfocitos en sangre periférica, médula ósea, ganglios linfáticos, bazo y otros tejidos.La LLC es el tipo de leucemia más frecuente en los individuos caucásicos y resulta raro en los asiáticos. En los países occidentales su incidencia es de alrededor de 3 casos nuevos por 100 000 habitantes y por año. En Europa y en Estados Unidos de América representa el 0,8 % de todas las neoplasias y cerca del 30 % de la totalidad de las leucemias. Su incidencia tiene una gran variación de acuerdo con el área geográfica, pues varía desde el 2,5 % de todas las leucemias en Japón hasta el 38 % en Dinamarca. Se presenta generalmente en personas de edad avanzada y es la más común después de los 50 años. Se ha señalado que sólo el 10 % de los enfermos tiene menos de esa edad en el momento del diagnóstico.1-4 Su incidencia depende de la edad, pues aumenta de 5,2 a 30,4 casos por 100 000 personas por encima de las edades de 50 y 80 años, respectivamente.5 La variedad más frecuente es la de linfocitos B, que es a la que nos referimos en el presente trabajo.
La etiología es desconocida. La LLC es la única leucemia del adulto en la que no se ha encontrado asociación entre factores de exposición, radiaciones ionizantes, agentes químicos o virus, y la aparición de la enfermedad. A favor de este criterio está el hecho de que no existen evidencias de que los japoneses supervivientes a las radiaciones producidas por la bomba atómica lanzada en su país hayan contraído un mayor riesgo de padecer LLC. Por otra parte, se han comunicado algunos casos en Jamaica de infección por HTLV-1 en pacientes con LLC, pero en ellos las células leucémicas no estaban infectadas por el virus, aunque sí sintetizaban anticuerpos contra la proteína p24 de la envoltura viral, lo que sugirió la hipótesis de que la LLC en estos casos podía resultar más bien como consecuencia de una posible respuesta inmune frente a la infección por este retrovirus que de una infección viral. Se ha señalado que aunque las células de la LLC pueden estar infectadas con el virus de Epstein-Barr no existen evidencias directas de que sea un agente causal. Sin embargo, existen ciertos factores relacionados con esta entidad: la epidemiología sugiere que el género y las características raciales influyen en el desarrollo de esta leucemia. Así, se conoce que la LLC es de 2 a 3 veces más frecuente en los hombres y su incidencia es menor en los países asiáticos y en México, pues se ha señalado que en este país latinoamericano su frecuencia es 6 veces menor que en las poblaciones caucásicas. La incidencia anual de LLC en Corea es sólo el 1,5 % de la que tiene los EE.UU. Un dato importante en este sentido es que entre los japoneses y chinos que emigran a América la incidencia de LLC se mantiene baja.1-4 Por otro lado, se han comunicado algunas familias con varios miembros afectados por LLC, por lo que se ha planteado la existencia de una base hereditaria, aspecto que no ha sido comprobado.6 Los estudios realizados en gemelos univitelinos con LLC sugieren la naturaleza adquirida de la enfermedad.2
Cuadro clínico
La LLC presenta diversas manifestaciones clínicas. Con mucha frecuencia, aproximadamente en la mitad de los casos, el diagnóstico se realiza en un individuo totalmente asintomático que se ha hecho un hemograma de rutina o en alguno que se hizo esta prueba por presentar síntomas de una enfermedad banal. En otras ocasiones, los primeros síntomas consisten en aparición de adenopatías, astenia y mal estado general. A diferencia de lo que sucede en los linfomas, la fiebre, sudoración y pérdida de peso no son habituales. Otras veces son las infecciones repetidas, virales o bacterianas, las que motivan la indicación de un hemograma que pone de manifiesto la enfermedad. Es mucho más raro que la primera manifestación de una LLC sea una anemia hemolítica autoinmune (AHAI).2,5El examen físico también es variable, pues puede ser negativo como sucede en algunos casos con enfermedad poco avanzada o bien mostrar adenopatías, hepatomegalia y esplenomegalia propias de la infiltración progresiva por los linfocitos leucémicos. En los casos con enfermedad poco avanzada se pueden encontrar pequeñas adenopatías localizadas, particularmente en la región cervical o supraclavicular. Sin embargo, a medida que la enfermedad progresa, las adenopatías se hacen más manifiestas y en ocasiones se presentan numerosas adenopatías simétricas indoloras generalizadas que pueden aumentar de tamaño y unirse en grandes masas, a veces notables en las regiones retroperitoneal y mesentérica. En esta situación, la palpación abdominal evidencia con frecuencia las grandes masas tumorales. Las adenopatías mediastínicas o la infiltración del anillo de Waldeyer son muy raras.
Aproximadamente en el 20-30 % de los casos se palpa una esplenomegalia. En general su tamaño es moderado, pero puede variar desde sólo una punta de bazo, que rebasa el reborde costal izquierdo durante la inspiración profunda, hasta una esplenomegalia masiva que en algunos pacientes llega a ocupar todo el hemiabdomen izquierdo. Aproximadamente en el 50 % de los enfermos se encuentra una hepatomegalia ligera. En los estadios más avanzados puede además producirse la infiltración de órganos no linfoides como la próstata, el hígado, en ocasiones con signos de hipertensión portal, el tubo digestivo, el pulmón, la pleura o el sistema nervioso central y más raramente de otros tejidos. A diferencia del dermotropismo que se observa en los síndromes linfoproli-ferativos de tipo T, en la LLC de tipo B la infiltración cutánea es rara y cuando se presenta, es más frecuente que sea reactiva que infiltrativa. En raras ocasiones se ha comprobado la coexistencia de LLC y síndrome nefrótico, por glomerulonefritis membranosa, que mejora cuando la enfermedad responde al tratamiento.2,5,7-9
Complicaciones
En la LLC pueden presentarse diferentes complicaciones. A medida que la enfermedad avanza aumentan las posibilidades de complicaciones secundarias al aumento de la masa tumoral, y es frecuente la aparición de trombocitopenia y anemia secundarias a insuficiencia medular. Muchos enfermos presentan en algún momento de su evolución un estado de inmunodeficiencia adquirida y con mayor frecuencia infecciones bacterianas (74 %), en gran parte de localización pulmonar o del tracto genitourinario, o infecciones virales (21 %), generalmente producidas por virus de tipo herpes. Estas últimas se han incrementado particularmente en los casos tratados con fludarabina. Con menor frecuencia se detectan infecciones micóticas; entre ellas se han comunicado meningitis criptocócica e histoplasmosis diseminada. Recientemente se ha planteado la posibilidad de instalación de micosis sistémicas, sobre todo producidas por Candida o Aspergilus. Se ha señalado que las infecciones constituyen la principal causa de mortalidad y de morbilidad en la LLC, pues llegan a producirse aproximadamente en el 80 % de los pacientes. La mortalidad de causa infecciosa, según diferentes estudios, está comprendida entre el 30 y el 50 %.10,11 Ese estado de inmunodeficiencia predispone a la aparición de segundas neoplasias. Cuando un paciente con LLC presente nuevos síntomas o toma del estado general, sin que para ello exista una explicación adecuada, debe pensarse en la posibilidad de una neoplasia asociada. Las segundas neoplasias generalmente son carcinomas cutáneos, del tubo digestivo o del pulmón. También se han comunicado casos de LLC asociados a algunas hemopatías mieloides, entre ellas leucemia mieloide crónica, policitemia vera, trombocitemia esencial y leucemia mieloblástica, y síndromes mielodisplásicos, pero sin que se haya establecido una relación causal entre los procesos.2,5,9Entre el 10 y el 30 % aproximadamente de las LLC se produce una transformación en un proceso linfoproliferativo más agresivo.9,12,13 La forma más frecuente de transformación es la prolinfocitoide, que tiene una incidencia del 15 al 30 %. En esta situación, la transformación se va expresando por un aumento gradual de los prolinfocitos en la LLC o en su forma mixta prolinfocítica (LLC/LPL), y desde el punto de vista clínico se asocia generalmente con un aumento de las adenopatías, esplenomegalia progresiva, anemia y trombocitopenia. Estas células que morfológicamente son prolinfocitos, generalmente mantienen las características inmunofenotípicas de la LLC, por lo que se consideran prolinfocitos con características intermedias, motivo por el cual con frecuencia se usa el término de «prolinfocitoides» para su denominación. Con mucha menor frecuencia, el aumento de los prolinfocitos se produce bruscamente; en esta situación a diferencia de la anterior, es usual que los prolinfocitos sean morfológica e inmunofenotípicamente idénticos a los de la leucemia prolinfocítica.
Alrededor del 10 % de las LLC pueden desarrollar un linfoma agresivo. La aparición de un linfoma difuso de células grandes constituye el denominado síndrome de Richter. En cerca de la mitad de los casos el linfoma surge a partir de la transformación del propio clon leucémico. Esta evolución al parecer comienza en los ganglios afectados por la LLC, aunque este síndrome puede manifestarse en órganos extranganglionares como el tubo digestivo o el pulmón.2 Las células de este linfoma son capaces de infiltrar la médula ósea y aparecer en la sangre periférica. La transformación hacia un síndrome de Richter se debe sospechar en aquellos enfermos que presenten fiebre de causa no identificada, pérdida de peso, adenopatías progresivas y aparición o intensificación de citopenias. Otros linfomas que con poca frecuencia se han encontrado asociados con la LLC son el linfoma de Burkitt, el histiocítico y la enfermedad de Hodgkin. En estos casos, el diagnóstico diferencial entre la enfermedad de Hodgkin y un linfoma de células grandes con células atípicas es muy difícil sobre una base puramente morfológica y generalmente se requiere de estudios inmunofenotípicos y moleculares para esclarecer la situación. En los casos de linfomas con células atípicas que semejan células de Reed-Sternberg, éstas presentan características diferentes, pues a diferencia de la clásica célula de Reed-Sternberg, son CD45+, CD15-, expresan antígenos B, como CD19, CD20 y CD22 y tienen reordenamiento de los genes de las inmunoglobulinas.12
La transformación en una leucemia linfoblástica es una complicación muy rara que se presenta en menos del 0,1 % de las LLC.14,15 Por otra parte, se han comunicado casos excepcionales de aparición de un mieloma múltiple, pero que en su mayoría no tiene relación con el clon leucémico.2
Trastornos inmunológicos
En la LLC se asocian frecuentemente trastornos de la regulación inmune y fenómenos de autoinmunidad. Las enfermedades autoinmunes que usualmente se presentan son la AHAI y la púrpura trombocitopénica de naturaleza inmune. Con una frecuencia mucho menor se observa una aplasia selectiva eritropoyética o una neutropenia secundarias a la producción de autoanticuerpos dirigidos contra las células progenitoras de esas líneas medulares. La prueba de Coombs resulta positiva en el 15 al 35 % de los casos, ya sea al diagnóstico o durante la evolución de la enfermedad, pero no siempre se acompaña de AHAI. Alrededor del 10 al 20 % de las LLC presentan AHAI en algún momento de su evolución. En estos casos se observan numerosos microesferocitos y policromatofilia en la extensión de sangre periférica, aumento de la cifra de reticulocitos y hay un incremento eritropoyético en la médula ósea. Los anticuerpos suelen ser calientes de tipo IgG.2,16,17Las causas del incremento de autoanticuerpos contra los eritrocitos y las plaquetas en la LLC, no están completamente esclarecidas. Se ha señalado que es posible que estos autoanticuerpos no sean producidos por las células B leucémicas, pues existen evidencias de que las cadenas ligeras y los isotipos de las cadenas pesadas de estos autoanticuerpos (generalmente IgG con cadenas ligeras k y l ) no están restringidas a las que presentan habitualmente las células B leucémicas (generalmente IgM o IgM/IgD y con sólo un tipo de cadena ligera). Por otra parte, se ha comprobado que la terapéutica inmunosupresora puede con frecuencia inducir la remisión del proceso autoinmune sin que se afecte significativamente la masa tumoral. Estas observaciones tomadas en su conjunto sugieren que estos autoanticuerpos no están directamente relacionados con las células malignas, sino que se producen por células B residuales no relacionadas con el clon leucémico.3
Frecuentemente, los anticuerpos existentes en la LLC reaccionan con autoantígenos principalmente con IgG. Estos autoanticuerpos se han denominado «polirreactivos», pues son anticuerpos que se pueden unir a 2 o más antígenos diferentes, usualmente con baja afinidad.3,16,17 Sin embargo, se ha sugerido que las células B de la LLC no son las que producen directamente los autoanticuerpos, sino que lo hacen de una forma indirecta actuando como una célula presentadora de antígenos unidos específicamente a sus inmunoglobulinas de superficie.3,18
En estos pacientes es frecuente la hipogammaglobulinemia; su prevalencia varía del 10 al 100 %, y se correlaciona con la duración de la enfermedad y el estadio en que se encuentra, pues es más frecuente en los pacientes con enfermedad avanzada.10,11 La Ig que está disminuida con mayor frecuencia es la IgM, seguida de la IgG y de la IgA. Entre el 5 y 10 % de los casos puede detectarse un componente monoclonal en el suero (frecuentemente IgM o IgG) que tiene las mismas características idiotípicas de las inmunoglobulinas de superficie de los linfocitos B. También se han descrito alteraciones del complemento y de la actividad fagocítica.11
En nuestro centro hemos encontrado una disminución de la actividad total de la vía clásica y la concentración de C1q y C4 en los estadios avanzados de la enfermedad. En estas etapas también se detectó la presencia de inmunocomplejos circulantes.19
La inmunidad celular también está manifiestamente alterada en la LLC. Existe un aumento de la cifra absoluta de los linfocitos T, aumento de la actividad de los linfocitos T supresores, disminución de los auxiliares y una inversión del índice CD4/CD8 en sangre periférica. Además está disminuida la actividad de las células citotóxicas naturales (NK), aunque éstas pueden estar en cantidad normal. También se conoce que puede haber disminución e incluso anergia a los antígenos usados en las pruebas cutáneas de hipersensibilidad retardada. Se ha comunicado en ocasiones una intensa disminución de los linfocitos CD4+, después del uso de los medicamentos análogos a las purinas, y también disminución de la formación de colonias celulares de tipo T.11,20
Morfología
Esta leucemia es considerada una enfermedad heterogénea teniendo en cuenta que su morfología celular no es siempre uniforme y que además puede presentar variaciones en sus características inmunofenotípicas, citogenéticas y moleculares. Por otra parte, existe gran variabilidad en su forma de presentación clínica y de evolución.Desde el punto de vista morfológico, la LLC típica está representada por una población monoclonal de linfocitos pequeños y no más del 10 % de linfocitos grandes, prolinfocitos o células atípicas en sangre periférica o médula ósea. Habitualmente se observan abundantes sombras de Gumprecht en la extensión de sangre periférica.
Se han señalado distintos subtipos de la enfermedad. Hace varios años se plantearon 2 variantes morfológicas además de la forma típica: una constituida por una población homogénea de linfocitos similares a los que se ven en las infecciones virales, cuyo citoplasma se adapta a la superficie de los hematíes, que se denominó de linfocitos activados, y otra en que existía una población de estas células junto a los linfocitos pequeños típicos. En la actualidad no se emplea esta clasificación. El grupo Franco-Americano-Británico (FAB) ha propuesto una clasificación de la LLC basada en la morfología de los linfocitos en sangre periférica: LLC típica donde más del 90 % de los linfocitos son pequeños y de aspecto maduro y las variantes consideradas de tipo mixto que incluyen el subtipo con aumento de prolinfocitos o células prolinfocitoides (LLC/LPL) en que el número de prolinfocitos está entre 11 y 54 %, y el subtipo llamado LLC atípica cuando existe heterogeneidad citomorfológica (linfocitos grandes, linfocitos hendidos, linfocitos granulares, inmunoblastos o eventualmente células linfoplasmocíticas), pero siempre con un número de prolinfocitos menor del 10 %.21,22
Inmunofenotipo
Las células leucémicas son linfocitos B que tienen un inmunofenotipo definido por 3 características principales: comparten antígenos B (expresión débil de los antígenos CD19, CD20, CD22 y reactividad habitual con CD23) con fuerte positividad del antígeno T CD5 en ausencia de otros marcadores T; expresan sólo una cadena ligera de las inmunoglobulinas (k o l ) y tienen una baja densidad de inmunoglobulinas de superficie (IgS) con una capacidad muy reducida de formación de casquetes en la superficie del linfocito. Estos elementos son adecuados para un diagnóstico preciso de la LLC y muy importantes para establecer el diagnóstico diferencial con otros síndromes linfoproliferativos crónicos.3 En la mayor parte de los casos, expresan IgM de superficie con o sin IgD, raramente expresan IgG, IgA o IgD, o pueden expresar alteraciones tipo talasémicas de las IgS.23 Prácticamente en todos los casos se encuentra inmunoglobulina citoplasmática, incluyendo aquéllos que no expresan IgS.Los linfocitos de la LLC forman rosetas espontáneas con los hematíes de ratón (rosetas M), que superan el 50 % cuando los linfocitos son tratados con neuraminidasa. Este es un marcador que resulta de gran utilidad. El grupo FAB lo ha propuesto dentro de los criterios de diagnóstico inmunológico de la LLC-B y ha planteado una cifra de al menos el 30 % para el diagnóstico.
Se ha señalado que casi todas las LLC expresan los antígenos CD5, DR, CD19, CD20 (a baja densidad), CD24, CD27, CD37, CD39, CD40, CD44, CD45RA, CDw75; muchas expresan también CD18, CD21 y CD23. Un número menor de casos expresan otros antígenos B. Los linfocitos de la LLC-B pueden en algunos casos coexpresar antígenos mielomonocíticos (CD11, CD13, CD14, Cd15, CD33), y en pocas ocasiones algunos antígenos T asociados (CD1, CD2, CD3, CD4, CD8).
Las células de la LLC-B expresan comúnmente bajos niveles de IgS al igual que de CD79a y Cd79b, que representan las moléculas accesorias del complejo constituido por el receptor inmunoglobulínico.2,3,9,24,25
Se ha indicado que las alteraciones en la expresión de las moléculas de adhesión influyen en los patrones de localización y diseminación hemoperiférica de los síndromes linfoproliferativos crónicos, por lo que su caracterización en los clones neoplásicos puede contribuir al diagnóstico y pronóstico de estos procesos. En diferentes síndromes linfoproliferativos se han observado alteraciones en la expresión de las moléculas de adhesión. Sin embargo, la presencia de diferentes patrones de expresión en varias muestras del mismo paciente, indica que la variabilidad entre las muestras puede depender de la etapa de maduración y activación, y aún de la función que desempeña determinada molécula de adhesión, lo que sugiere que el grado de expresión de estas moléculas en una determinada muestra está en dependencia del estado en que las células se encuentren en ese preciso momento.
En un trabajo reciente se analizó la expresión de diferentes moléculas de adhesión, que incluían las familias de las integrinas, selectinas y la superfamilia de las inmunoglobulinas, en varios síndromes linfoproliferativos crónicos de tipo B.26 Se pudo apreciar que en la LLC las células expresaban una intensidad muy tenue de todas las integrinas b, en comparación con los controles y con otros procesos linfoproliferativos, dato que coincide con lo comunicado en otros estudios.27,28 Las integrinas b1, tanto CD49d como CD29 se expresaban muy débilmente, pero se observó que la CD49d aumentaba según se incrementaba la masa tumoral, aunque su expresión se mantenía por debajo de la de los linfocitos normales, mientras que la de CD29 era significativamente mayor en los pacientes con esplenomegalia. El análisis de las integrinas b2 mostró que había una expresión muy baja de CD11a/CD18, así como de CD11b. Sin embargo, la CD11c se expresaba fuertemente y la expresión de CD11a era mayor en las etapas avanzadas de la LLC.
El determinante CD54, que es miembro de la superfamilia de las inmunoglobulinas y representa al ligando de CD11a/CD18, sólo se manifestó en menos del 20 % de las células, aunque su expresión parece ser mayor en los estadios más avanzados de la enfermedad. De las selectinas estudiadas, la representada por CD62L estaba disminuida y la expresión de CD44 era similar a la de los linfocitos normales y a la de las células de otros síndromes linfoproliferativos. De acuerdo con estos datos, la LLC se podría caracterizar por la baja expresión de las integrinas b 1, b 2 la correlación positiva que parece existir entre la intensidad de la expresión de CD49d/CD29, CD11a y CD54 y el volumen de la masa tumoral, mientras que la expresión de CD29 parece ser mayor en los pacientes con esplenomegalia. La baja expresión de CD11a/CD18 en la LLC podría contribuir al diagnóstico diferencial con el linfoma linfocítico de células pequeñas que siempre es positivo para este marcador. El patrón de expresión del CD11a/CD18 en estos 2 procesos linfoproliferativos, semejantes en su citomorfología y en su inmunofenotipo, pudiera explicar la retención de las células del linfoma en los órganos linfoides y su baja diseminación hemoperiférica.26-28
Estudio citogenético
Mediante las técnicas de bandas G se han puesto en evidencia anomalías cromosómicas aproximadamente en el 50 % de las LLC, que según algunos son más frecuentes en los casos en fases avanzadas.29 Se ha señalado que la frecuencia de estas anomalías es mayor en las LLC clasificadas como de tipo mixto. Dentro de las alteraciones citogenéticas se ponen de ma-nifiesto solamente anomalías únicas en más de la mitad de los casos, mientras que sólo aproximadamente en el 10 al 20 % se observan cariotipos complejos. El análisis de una amplia casuística mostró que la alteración más frecuente estaba representada por la trisomía 12, seguida de anomalías estructurales 13q y en orden decreciente 14q, anomalías del cromosoma 11, del cromosoma 6 y de los cromosomas 17, 1, 8, 7 y 18.29-31Un paso importante en el estudio citogenético de las LLC ha sido la aplicación de las técnicas moleculares, en particular el análisis de la hibridación in situ mediante sondas marcadas con fluorescencia (FISH), que ha dado origen a la citogenética molecular. Esto ha sido un logro importante si tenemos en cuenta el alto número de casos con cariotipo normal por las técnicas tradicionales en una enfermedad de proliferación lenta y donde existía la posibilidad de que los mitógenos usados in vitro pudiesen estimular las células T o los linfocitos B policlonales. Con el empleo de esta técnica hoy es posible demostrar una serie de anomalías cromosómicas con una frecuencia mayor a la sospechada. Así, se ha visto una incidencia de trisomía 12 del 20 % con la ténica de FISH, que es aproximadamente el doble de la que se aprecia con los métodos citogenéticos convencionales. Se ha señalado que esta trisomía se asocia con un aumento de los prolinfocitos (LLC/LPL) y una tendencia a desarrollar una enfermedad más agresiva. Sin embargo, el significado patogénico de esta alteración permanece oscuro y tampoco está bien esclarecido si es evidencia de un peor pronóstico.29,30
La reciente demostración de deleciones del cromosoma 13q12-14 aproximadamente del 45 al 80 % de los pacientes ha abierto nuevas posibilidades de estudio en la LLC. Se ha observado una deleción de este cromosoma localizada en la banda 13q12 en una región de alrededor de 500 kb que comprende el área del gen BCRA2, así como otras deleciones independientes que ocurren al nivel del 13q14, cercanas al gen de susceptibilidad al retinoblastoma (RB). Algunas de estas delecciones ocurren en ambos alelos del cromosoma 13. Actualmente se investiga si en esta región existen uno o más oncogenes supresores que se asocien con la patogenia de la LLC.31
En fecha reciente se ha comunicado que las alteraciones estructurales, particularmente deleciones, del brazo largo del cromosoma 11(11q) se presentan en el 20 % de las LLC, específicamente en la región 11q22-23 y se ha señalado que la deleción 11q se correlaciona fuertemente con un mal pronóstico e identifica un subtipo de LLC que se caracteriza por presentarse en estadios clínicos avanzados y por acentuadas linfoadenopatías periféricas e internas.31-32
Biología molecular
La aplicación de las técnicas moleculares basadas en la hibridación del DNA ha permitido profundizar los conocimientos relacionados con la biología de las células leucémicas en la LLC. El estudio del reordenamiento de los genes de las inmunoglobulinas, que incluye el de las cadenas pesadas y ligeras, ha posibilitado la confirmación de la naturaleza clonal de esta enfermedad, y ha proporcionado un método más preciso para la identificación de la enfermedad mínima residual. La reciente introducción de la técnica de reacción en cadena de la polimerasa, procedimiento muy sensible, que es capaz de identificar una célula leucémica entre 105-106 células normales, ha hecho posible ampliar el concepto de remisión de la enfermedad hasta el grado de remisión molecular.33-35Hasta el momento no se ha demostrado la existencia de una alteración genética molecular asociada con el desarrollo de la LLC. La participación del oncogén BCL-2 en la génesis de la LLC ha sido objeto de múltiples estudios. Se ha señalado que en esta leucemia los linfocitos se acumulan en gran parte debido a una inhibición de la apoptosis secundaria a la sobreexpresión del gen BCL-2, aún cuando el reordenamiento de este gen es poco frecuente en esta enfermedad. En la LLC se han observado altos niveles de la proteína BCL-2 hasta en más del 85 % de los casos.36 Sin embargo, en un estudio reciente el gen estaba reordenado en sólo el 12 % de los enfermos.37 Estos datos hacen pensar en diversos mecanismos genéticos, hasta el momento sin esclarecer, que pueden intervenir en la desregulación de este oncogén en la LLC. Ningún otro oncogén comprometido en la patogenia de otros procesos linfoproliferativos B, como el BCL-1, BCL-6, PAX-5 y MXC, se altera primariamente en la LLC.
Las variaciones de los reguladores del ciclo celular: p53, p15, p16, se asocian generalmente con formas clínicas agresivas o con la transformación en linfomas de células grandes. La inactivación del gen oncosupresor p53 se ha encontrado en un pequeño porcentaje de pacientes (10-20 %), pero esta modificación probablemente representa eventos moleculares asociados con la progresión o la transformación tumoral.38 Por otra parte, no se han comunicado aberraciones recurrentes de la p15 y la p16 en los procesos linfoproliferativos crónicos de tipo B.31,39
Algunos trabajos habían sugerido que el gen RB estaba comprometido en la patogenia de la LLC, teniendo en cuenta las frecuentes deleciones que se observan en el locus 13q14 correspondiente a su localización. Estudios posteriores han puesto en evidencia que el RB raramente resulta inactivado en la LLC y que por lo general, las deleciones se producen alejadas del gen RB en sentido distal.31 Al parecer, las deleciones que ocurren en la región 13q14 representan aberraciones clonales tempranas en la LLC y sugieren fuertemente la presencia de un gen oncosupresor, cuya pérdida o inactivación puede ser crucial para el desarrollo de la LLC. Para tratar de identificar este posible gen se han realizado algunos estudios moleculares que han puesto en evidencia una mínima región de deleción (MRD) que es común en todos los casos de LLC que tienen deleciones 13q14. Esta región está ubicada teloméricamente al gen RB y no incluye a éste, pero contiene al parecer nuevos genes que representan candidatos al gen oncosupresor asociado con la LLC. Por otra parte, se ha comprobado mediante sondas moleculares aplicadas en esta MRD, que aunque la inmensa mayoría de las delecciones 13q14 no se aprecian con las técnicas citogenéticas convencionales, molecularmente pueden detectarse en más del 50 % de las LLC. Hasta el momento no se ha podido identificar el posible gen oncosupresor asociado con la LLC, pero las investigaciones se mantienen en este sentido, pues su identificación sería de gran importancia para el mejor conocimiento del desarrollo de este tipo de leucemia.31,40,41
Origen de la célula leucémica
La LLC se caracteriza por la acumulación de linfocitos B CD5+ y por una alta frecuencia de generación de fenómenos autoinmunes. Se ha señalado que estas características son el reflejo de las presentes en los linfocitos B CD5 normales. Esto último constituye una pequeña subpo-blación de linfocitos B existente en los órganos linfáticos y la sangre de los adultos normales y en los tejidos linfoides del feto.24,42,43En el adulto, los linfocitos B CD5+ se pueden encontrar como elementos sésiles localizados en los bordes de los centros germinativos y en las zonas del manto que rodean los centros germinativos de los folículos secundarios. Sin embargo, las características biológicas de las células CD5+ de la LLC y las de los linfocitos B normales CD5+ no son exactamente iguales, pues aunque existen múltiples semejanzas también hay algunas diferencias.18
Los linfocitos B normales CD5+ entre otros caracteres, expresan generalmente niveles normales de inmunoglobulinas de superficie IgM, IgD, y excepcionalmente IgG, además pueden producir espontáneamente autoanticuerpos naturales IgM polirreactivos de baja afinidad y frecuentemente del tipo del factor reumatoideo, expresan idiotipos con reactividad cruzada e inmunoglobulinas con escasas o ninguna mutación somática, forman rosetas espontáneas con eritrocitos de ratón, expresan algunos antígenos mielomonocíticos y bajos niveles de CD20, se estimulan por el virus de Epstein-Barr (VEB) y expresan regularmente la proteína C-MYC. Estas células mantienen un adecuado sistema de transmisión de señales, con respuesta normal a la acción del Ca++ y del funcionamiento del mecanismo de intercambio Na+/H+, que es un paso fundamental en la transmisión de señales provenientes de muchos factores de crecimiento y de mitógenos.3,18,24,42,43
Por otra parte, los linfocitos B CD5+ de la LLC, aunque conservan muchas de estas características, se diferencian de los normales en los siguientes rasgos principales: expresión de bajos niveles de inmunoglobulinas de superficie, lo que es un elemento distintivo de la LLC, resistencia a la transformación por el VEB aún cuando ellos expresan el receptor celular para el virus (CD21), disminución de la transmisión de señales intracelulares que los incapacita para responder a diversos estímulos, marcada reducción de la función de intercambio Na+/H+ y de la estimulación por el Ca++.44-47 Las células de la LLC expresan altos niveles de la proteína BCL-2 usualmente en ausencia de un reordenamiento del gen, lo que las hace resistentes a la apoptosis y pueden facilitar su acumulación progresiva, y además, baja expresión de Fas y ausencia de la proteína C-MYC. También se ha señalado que ellas se encuentran detenidas en la fase Go de su ciclo celular.18
Aunque los linfocitos B normales CD5+ y los de la LLC comparten muchas características comunes, no tienen exactamente el mismo fenotipo, pues existen entre ellos muchos factores diferenciales. Por esto se ha sugerido que la contrapartida normal del linfocito de la LLC puede ser una subpoblación de los linfocitos normales CD5+ que durante su desarrollo fisiológico haya adquirido un fenotipo similar al de los de la LLC. Sobre esta base, se ha planteado la hipótesis de que la LLC sea una enfermedad ocasionada por el acúmulo de una subpoblación de linfocitos B CD5+ «anérgicos», que se encuentran fisiológicamente comprometidos en la producción de autoanticuerpos naturales. En este caso, el término anergia se usa para caracterizar a los linfocitos CD5+ que son funcionalmente inactivos después que han recibido una estimulación subóptima por autoantígenos. Normalmente, después que estas células son anergizadas por la exposición a autoantígenos solubles, se produce una disminución de la expresión de las inmunoglubulinas de superficie, tal como se ve en la LLC y adquieren además otras características peculiares de esta leucemia.3,18
De acuerdo con esa hipótesis, la transformación del linfocito B normal CD5+ en el linfocito de la LLC se realiza a través de varios pasos sucesivos (fig). Inicialmente los linfocitos B normales CD5+ se transformarían en linfocitos CD5+ anérgicos, mediante una exposición subóptima a autoantígenos solubles, los que en etapas posteriores por estímulos aún desconocidos sufrirían la transformación maligna, mediante la cual adquirirían una serie de propiedades como la incapacidad de responder al VEB, disminución funcional del intercambio Na+/H+, disminución de la transmisión de señales intracelulares, entre otras alteraciones que favorecen su acúmulo en la fase Go del ciclo celular.18,44-47
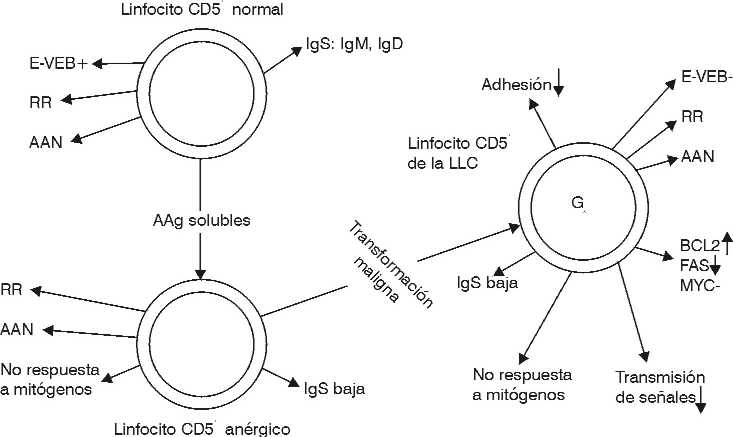
Con toda seguridad, el extraordinario avance alcanzado en los últimos años en el campo científico-técnico, y en particular en el área de la biología molecular, permitirá en un futuro cercano esclarecer muchas de las interrogantes que aún se mantienen referentes al desarrollo de la célula leucémica de la LLC y los factores genéticos que en él intervienen.
Summary
Chronic lymphocytic leukaemia (CLL) is the commonest type of leukaemia among Caucasian individuals. Its incidence varies a lot according to the geographical area. The most frequent variety is that of B lymphocytes described in this paper. Its etiology is unknow. Generally, the CLL may have a wide range of clinical manifestations and different complications. In approximately 10 to 30 % of the cases it is observed a change to a more aggressive lymphoproliferative process. The CLL is a heterogenous disease in this cytomorphological aspect and may present variations in its immunophenotypical, cytogenetic and molecular characteristics. The modern molecular techniques have demostrated a higher incidence of cytogenetic disorders than the traditional methods. The most frequent disorders were trisomy 12 and deletions of 11q and of 13q12-14. The molecular studies have not been able to identify an oncogen associated with the development of CLL. However, an active research in being carried out in this sense.Subject headings: LEUKEMIA, B-CELL, CHRONIC.
Referencias Bibliográficas
1. Montserrat E, López-Kartpovitch. Tratamiento de la leucemia linfocítica crónica, prolinfocítica y otros síndromes linfoproliferativos. En: Ruiz-Argüelles GJ, San Miguel JF, eds. Actualización en leucemias. México, DF: Editorial Médica Panamericana, 1996:107-13.
2. Montserrat E. Leucemia linfática crónica: clínica, pronóstico y terapia. En: López Borrasca. Enciclopedia iberoamericana de Hematología, Salamanca: Universidad de Salamanca, 1992; vol 2:310-22.
3. Rai KR, Kipps TJ, Barlogie B. Chronic lymphocytic leukemia and myeloma: update on the biology and management. Hematology 1996. Education Program American Society of Hematology, Orlando, 1996:62-73.
4. Brincker H. Population-based age and sex-specific incidence rates in the 4 main types of leukemia. Scand J Haematol 1982;29:241-9.
5. Faguet GB. Chronic lymphocytic leukemia: an updated review. J Clin Oncol 1994;12:1974-90.
6. Neuland CY, Blattner WA, Mann DL, Fraser MC, Tsai S, Strong DM. Familial chronic lymphocytic leukemia. JNCI 1983; 71:1143-50.
7. Worning AM, Dragsted J. Chronic lymphocytic leukemia diagnosed through histological evaluation of resected prostatic tissue. Urol Int 1985;40:292-3.
8. Warin AP, Roberts MM. Chronic lymphocytic leukaemia with cutaneous involvement. Clin Exp Dermatol 19789;4:241-6.
9. Montserrat E, Rozman C. Síndromes linfoproliferativos crónicos de expresión leucémica. En: Rozman C. Medicina interna, 12a ed Barcelona: Ediciones Doyma, 1992; vol 2: 1682-91.
10. Molica S. Infections in chronic lymphocytic leukemia: risks factors and impact on survival and treatment. Leuk Lymph 1994;13:203-14.
11. Nosari A, Santoleri L, Tedeschi A, Crugnola M, Coluccia P, Gini G, et al. Le complicanze infettive nella leucemia linfatica cronica. Sem Ematol 1997;5:103-14.
12. Faramarz N. Pathology of bone marrow. 2 ed. Baltimore: Williams and Wilkins, 1998:262-4.
13. Melo JV, Catovsky D, Galton DAG. The relationship between chronic lymphocytic leukaemia and prolymphocityc leukaemia. II. Patterns of evolution of «prolymphocytoid» transformation. Br J Haematol 1986;64:77-86.
14. Laurent G. Gourdin MF, Flandrin G, Kuhlein E, Pris J, Reyes F. Acute blast crisis in a patient with CCL: immunoperoxidase study. Acta Haematol 1981;65:60-6.
15. Miller ALC, Habershaw JA, Dhaliwhal HS, Lister TA. Chronic lymphocytic leukaemia presenting as a blast cell crisis. Leukem Res 1984;8:905-12.
16. Koeffler HP, Tesler AS, Golde DW. Chronic lymphocytic leukemia. En: Haskell CM, ed. Cancer treatment. 2 ed. Philadelphia: WB Saunders, 1985:729-35.
17. Kipps TJ, Carson DA. Autoantibodies in chronic lymphocytic leukemia and related systemic autoimmune diseases. Blood 1993;81:2475-87.
18. Caligaris-Cappio F. B-chronic lymphocytic leukemia: a malignancy of anti-self B cells. Blood 1996;87:2615-20.
19. Villaescusa R, Borrego I, Merlín J, Hernández P. Estudio seriado del sistema complemento e inmunocomplejos circulantes en la leucemia linfoide crónica. Rev Cubana Hematol Immunol Hemoter 1989;5:37-44.
20. Hernández P. Leucemia linfoide crónica. Estudio inmunológico. Rev Cubana Med 1981;20:424-35.
21. Sherdlow SH, Hurtubise PE. Laboratory evaluation of chronic lumphocytic leukemia. En: Bick RL, Hematology. Clinical and Laboratory practice. Vol 1. Baltimore: Mosby, 1993; vol 1:815-29.
22. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, et al. The French-American-British (FAB) cooperative group. Proposals for the classification of chronic (mature) B and T lumphoid leukaemias. J Clin Pathol 1989;42:567-84.
23. Martínez G, Ferreira R, Hernández A, Hernández P, Cruz C, Fernández O, et al. The molecular heterogeneity of chronic lymphocytic leukemia: Futher data. Haematologica 1988;73:169-72.
24. Bernasconi C, Pagnucco G, Caberlon S. Immunobiologia della LLC e dellHCL. Sem Ematol 1997;5:15-28.
25. Duque R, Orfao A. Utilidad del inmunofenotipo en el diagnóstico y clasificación de las leucemias crónicas. En: Ruiz-Argüelles GJ, San-Miguel eds. Actualización en leucemias. México, De: Editorial Médica Panamericana, 1996:89-96.
26. Lucio P, Faria M, Pinto A, Lopes M, Ribeiro M, Marques R, et al. Expression of adhesion molecules in chronic B-cell lumphoproliferative disorders. Haematologica 1998;83:104-11.
27. Baldini LG, Cro IM. Structure and function of VLA integrins: differential expression in B-cell leukemia/lymphoma. Leuk Lymph 1994;12:197-203.
28. Csanaky G. Matutes E, Vass JA, Morilla R, Catovsky D. Adhesion receptors on peripheral blood leukemic B cells. A comparative study on B cell chronic lymphocytic leukemia and related lymphoma/leukemias. Leukemia 1997;11:408-15.
29. Castoldi G, Cuneo A, Piva N, Roberti MG, Bigoni R, Bardi A. Citogenetica e biologia, molecolare della LLC e della HLC. Sem Ematol 1997;5:29-44.
30. Fegan C, Robinson H, Thompson P, Whittaker JA, White D. Karyotypic evolution in CCL: identification of a new subgroup of patients with deletions of 11q and advanced or progressive disease. Leukemia 1995;9:2003-8.
31. Catovsky D. The search for genetic dues in chronic lymphocytic leukemia. Hematol Cell Ther 1997; (Suppl 1): 39:S5-S11.
32. Döhner H, Stilgenbauer S, James MR, Benner A, Weilguni T, Bentz M, et al. 11q deletions identify a new subset of B-cell chronic lymphocytic leukemia characterized by extensive nodal involvement and inferior prognosis. Blood 1997;89:2516-22.
33. Brugiatelli M, Callea V, Morabito F, Oliva B, Di Celle PF, Fierro MT, et al. Immunologic and molecular evaluation of residual disease in B-cell chronic lymphocytic leukemia patients in clinical remission phase. Cancer 1989;63:1979-84.
34. Lenormand B. Bizet M, Fruchart C, Tilly H, Daliphard S, Thouret F, et al. Residual disease in B-cell chronic lymphocytic leukemia patients and prognostic value. Leukemia 1994;8:1019-26.
35. Esteve J, Villamor N, Colomer D, Bosch F, López-Guillermo A, Rovira M, et al. Hematopoietic stem cell transplantation in chronic lymphocytic leukemia: a report of 12 patients from a single institution. Ann Oncol 1998:9:167-72.
36. Reed JC. BCL2 family proteins: role in deregulation of apoptosis and chemoresistance in B-CLL. Hematol Cell Ther 1997;39(Suppl 1):S22-4.
37. Merup M, Spasokoukotskaja T, Einhom S, Edvard Smith CI, Gahrton G, Juliusson G. BCL-2 rearrangements with breakpoints in both vcr and mbr in non-Hodgkins lymphomas and chronic lymphocytic leukaemia. Br J Haematol 1996;92:647-9.
38. Cobo F, Pinyol M, Bosch F, Hernández LL, Hernández S, Jares P, et al. Analysis of ciclin-dependent kinase inhibitors (CDKI) and p53 in chronic lumphocytic leukaemia (CLL): mutations of p53 and deletions of p16 INK 4A genes are associated with Richters syndrome (RS). [abstract] Hematol Cell Ther 1997;39(Suppl 1):S65.
39. Ajchenbaum-Cymbalista F, Delmer A. Cell cycle regulation in chronic lymphoproliferative disorders. ISH-EHA Combined Haematology Congress, 4-8 July, 1998. Amsterdam: Educational Program Book, 1998:120-5.
40. Dalla-Favera R. Oncogenesis in CLL. [abstract] Hematol Cell Ther 1997;(Suppl 1):S54.
41. Stilgenbauer S, Nickolenko J, James MR, Litters A, Wilhelm J, Bullinger L, et al. Molecular cytogenetic characterization of 13q 14 and 11q22-q23 aberrations in B-cell chronic lymphocytic leukaemia. [abstract]Hematol Cell Ther 1997;(Suppl 1):39S69.
42. Caligaris-Cappio F, Gobbi M, Bofill M, Janossy G. Infrequent normal B lymphocytes express features of B-chronic lymphocytic leukemia. J Exp Med 1982;155:623-8.
43. Burastero SE, Casali P. Characterization of human CD5 (Leu-1, OKT1)+ B lymphocytes and the antibodies they produce. Contrib Microbiol Immunol 1989;11:231-62.
44. Carlsson M, Totterman TH, Matsoon P, Nilsson K. Cell cycle progression of B-chronic lymphocytic leukemia cells induced to differentiate by TPA. Blood 1988;71:415-21.
45. Lankester AC, Schijndel GM van, Schoot CE van der, Oers MH van, Noesel CJ van, Lier RA van. Antigen receptor nonresponsiveness in chronic lymphocytic leukemia cells. Blood 1995;86:1090-1.
46. Ghigo D, Gaidano G, Treves S, Bussolino F, Pescarmona G, Caligaris-Cappio F, et al. Na+/H+ antiporter has different properties in human B lymphocytes according to CD5 expression and malignant phenotype. Eur J Immunol 1991;21:583-8.
47. Grinstein S, Rothstein A. Mechanisms of regulation of the Na+/H+ exchanger. J Membr Bio 1986;90:1-2.Recibido: 7 de diciembre de 1998. Aprobado: 9 de diciembre de 1998.
Dr. Porfirio Hernández Ramírez. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba.
Teléf. (537)578268. Fax(537)338979.e-mail:ihidir@hemato.sld.cu

