










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.18 n.3 Ciudad de la Habana sep.-dic. 2002
Artículos Originales
Estudio de algunos parámetros hematológicos, de la función hepática y renal en niños con drepanocitosis
Dr. Sergio A. Machín García,1 Dr. Luis E. Pérez Ulloa,1 Dra. Tania García Peralta,1 Dra. Eva Svarch1 y Dra. Maura Wade Mateo2
Resumen
Se realizó un estudio longitudinal retrospectivo de algunos parámetros hematológicos y de la función hepática y renal en 88 niños con drepanocitosis, 35 del sexo femenino y 53 del masculino; 63 con anemia drepanocítica y 25 con hemoglobinopatía SC, seguidos entre enero de 1986 y diciembre de 1997 en el Instituto de Hematología e Inmunología, con el objetivo de estudiar posibles alteraciones y su relación con la edad, sexo y el tipo de hemoglobinopatía. Se analizó el promedio anual de la hemoglobina, reticulocitos, leucocitos, plaquetas y los valores de alaninoamino transferasa (ALT), aspartato transferasa, fosfatasa alcalina sérica (FAS) y creatinina por año, agrupados en grupos etáreos (de 2 a 5, de 6 a 8, de 9 a 11 y de 12 a 15 años). La hemoglobina y reticulocitos mostraron diferencias significativas entre las hemoglobinopatías ( p< 0,05). La función hepática y renal estuvo dentro de los parámetros normales, pero en la anemia drepanocítica se observó un aumento gradual con la edad de la ALAT, FAS y creatinina ( p< 0,05).
DeCS: ANEMIA HEMOLITICA; ESTUDIOS LONGITUDINALES; FACTORES DE EDAD.
La drepanocitosis comprende un grupo de anemias hemolíticas crónicas que incluyen a la anemia drepanocítica (AD), la hemoglobinopatía SC (HSC) y la S/b talasemia (S/btal). Se caracteriza por la presencia de una hemoglobina anormal, la HbS, que es el resultado de una mutación en la globina b en la cual el ácido glutámico es sustituido por la valina.1 Esta mutación se produjo en zonas donde la malaria es endémica como en África, Arabia y la India, en las que los individuos heterocigóticos (AS) para la enfermedad tuvieron alguna ventaja selectiva con respecto a los individuos sanos (AA). De esta forma se perpetuó el gen mutado, que se difundió después a otras regiones en el mundo.2,3
La incidencia de la drepanocitosis en Cuba es alta.4 El evento primario en la fisiopatología de la drepanocitosis es la polimerización de la HbS en estado desoxigenado, con la formación de un gel extremadamente viscoso. La gelificación distorsiona el hematíe y disminuye su flexibilidad. Estas células tiene una sobrevida acortada, lo que produce una anemia hemolítica crónica. Los glóbulos rojos con HbS obstruyen los pequeños capilares en la microcirculación y ocasionan una oxigenación deficiente de los tejidos, con el consiguiente daño orgánico.
La severidad del cuadro clínico influye en la morbiletalidad, posiblemente determinado por factores genéticos, socioeonómicos y culturales.2-5
El cuadro clínico de la drepanocitosis es muy variable entre un paciente y otro, y en un mismo paciente en diferentes etapas de la vida.6,7 El pronóstico de la enfermedad ha mejorado notablemente, y el promedio de vida en la actualidad es de 42 años en los hombres y de 48 en las mujeres.8
Desde los primeros años de la vida, la anemia crónica, la oclusión vascular aguda a repetición, la oclusión vascular crónica constante y subclínica, y a veces la sobrecarga de hierro, conducen a la alteración permanente e irreversible de los órganos,9,10 pero la edad en que comienzan estas alteraciones aún no se conoce.11
El objetivo de este trabajo fue estudiar posibles alteraciones de algunos parámetros de laboratorio en el niño con drepanocitosis y su relación con la edad, sexo y el tipo de hemoglobinopatía.
Métodos
Los pacientes se estudiaron en las visitas de rutina a la Consulta de Hemoglobinopatías de nuestro instituto. Se excluyeron por su escaso número los pacientes S/b tal, los niños menores de 2 años de edad, y los que tuvieron un seguimiento menor de 2 años o irregular.
Se establecieron 4 grupos de edades: 2-5, 6-8, 9-11 y 12-15 años.
Las determinaciones de hemoglobina (Hb), reticulocitos, leucocitos y plaquetas se realizaron cada 3 meses, según los métodos habituales, y las de alaninoamino transferasa (ALT), la aspartato transferasa (AST), fosfatasa alcalina sérica (FAS) y creatinina, una vez al año. Todas las investigaciones se hicieron con el paciente en estado basal, es decir, libre de síntomas y signos y sin transfusión en los 3 meses previos al estudio.
El análisis estadístico se realizó de acuerdo con el tipo de hemoglobinopatía, grupos de edades y sexo, por medio del cálculo de frecuencias de las variables cualitativas. Para las variables cuantitativas se calculó la media (![]() ) y la desviación estándar (DE). Se aplicó el análisis de varianza (ANOVA) o la prueba de Kruskal-Wallis12 para comparar los promedios. El nivel de significación fue del 5 % en todos los casos.
) y la desviación estándar (DE). Se aplicó el análisis de varianza (ANOVA) o la prueba de Kruskal-Wallis12 para comparar los promedios. El nivel de significación fue del 5 % en todos los casos.
Resultados
Se estudiaron 88 niños con drepanocitosis entre enero de 1986 y diciembre de 1997, 35 del sexo femenino y 53 del masculino; 63 con anemia drepanocítica y 25 con HSC. La Hb fue más baja y los reticulocitos más elevados en la AD, con diferencias significativas con respecto a la HSC (p < 0,05). La media de leucocitos y plaquetas se encontró más elevada en la AD, sin diferencia significativa con la HSC. La ALT, AST, FAS y creatinina fueron similares en ambas hemoglobinopatías (tabla 1).
Tabla 1. Datos de laboratorio en la drepanocitosis
| Parámetro | Anemia drepanocítica | Hemoglobinopatía SC | ||
| Media | DE | Media | DE | |
| Hb (g/dL)* | 7,9 | 1,1 | 10,3 | 1,1 |
| Reticulocitos (x 10-3)* | 139 | 54,9 | 71,3 | 55,3 |
| Leucocitos (x 109/L) | 12,1 | 5 | 10,3 | 10,7 |
| Plaquetas (x 109/L) | 337,6 | 142,4 | 317,1 | 83,6 |
| ALT (UI) | 5 | 3,4 | 5 | 3,6 |
| AST (UI) | 5,7 | 2,8 | 6 | 4,5 |
| FAS (UI) | 5,3 | 2,8 | 5,4 | 2,5 |
| Creatinina (µmol/L) | 75,4 | 28,4 | 77,5 | 34,5 |
*p < 0,05.
Se observó menos anemia y reticulocitosis en el sexo femenino en la AD, con diferencias significativas (p < 0,05). El resto de los parámetros estudiados no presentaron variaciones importantes (tabla 2).
Tabla 2. Parámetros de laboratorio por sexo en la anemia drepanocítica y en la hemoglobinopatía SC
| Parámetro | Anemia drepanocítica | Hemoglobinopatía SC | ||
| Femenino | Masculino | Femenino | Masculino | |
| Hb (g/dL)* | 8,1* | 7,8 | 10,5 | 10,2 |
| Reticulocitos (x 10-3)* | 126,8* | 146,9 | 52,4 | 75,7 |
| Leucocitos (x 109/L) | 12,1 | 12,2 | 13 | 9,9 |
| Plaquetas (x 109/L) | 327,4 | 344,2 | 319,5 | 315,9 |
| ALT (UI) | 4,8 | 6,2 | 4,5 | 5,3 |
| AST (UI) | 4,4 | 4,9 | 5,7 | 6,1 |
| FAS (UI) | 5,4 | 5,2 | 5,7 | 5,2 |
| Creatinina (µmol/L) | 74,1 | 76,2 | 73,9 | 74,1 |
*p < 0,05.
En la HSC, la Hb y los leucocitos presentaron valores más elevados y los reticulocitos fueron más bajos en el sexo femenino, pero sin significación estadística. Las plaquetas, ALT, AST, FAS y creatinina no mostraron diferencias (tabla 2).
En las tablas 3 y 4 se presentaron los estudios hematológicos por grupos de edades en la AD y en la HSC, sin diferencias entre los grupos.
Tabla 3. Parámetros hematológicos por grupos de edades en la anemia drepanocítica
| Grupo de edades | ||||||||
| Parámetro | 2-5 | 6-8 | 9-11 | 12-15 | ||||
| Media | DE | Media | DE | Media | DE | Media | DE | |
| Hemoglobina (g/dL) | 8 | 1,2 | 7,8 | 1 | 8 | 1 | 7,9 | 1,2 |
| Reticulocitos (x 10-3) | 114 | 60,1 | 139 | 49,8 | 128,6 | 50,4 | 139,9 | 55,9 |
| Leucocitos (x 109/L) | 11,8 | 2,9 | 12,5 | 3,1 | 11,5 | 2,4 | 13,4 | 11,7 |
| Plaquetas (x 109/L) | 330,7 | 96,6 | 340,6 | 76,2 | 348,7 | 271,4 | 340,3 | 75,1 |
Tabla 4. Parámetros hematológicos por grupos de edades en la hemoglobinopatía SC
| Grupo de edades | ||||||||
| Parámetro | 2-5 | 6-8 | 9-11 | 12-15 | ||||
| Media | DE | Media | DE | Media | DE | Media | DE | |
| Hemoglobina (g/dL) | 10,3 | 1,4 | 10,1 | 0,9 | 10,4 | 1 | 10,6 | 0,7 |
| Reticulocitos (x 10-3) | 67,7 | 37,4 | 75,4 | 63,7 | 72,5 | 63 | 68,7 | 70,7 |
| Leucocitos (x 109/L) | 11,7 | 16 | 10,3 | 2,4 | 9,4 | 2,1 | 14,4 | 16,4 |
| Plaquetas (x 109/L) | 330,3 | 80,2 | 324,1 | 90 | 296,1 | 81,8 | 290,4 | 67,5 |
Los resultados de la función hepática en la AD se muestran en la figura 1, donde se aprecia que la ALT y la FAS se encuentran dentro de límites normales, pero con tendencia a aumentar con la edad, con significado estadístico (p < 0,05), la AST presentó una distribución normal.
Los resultados de los estudios de la función hepática en la HSC estuvieron dentro de límites normales en todas las edades, sin distribución definida ni diferencias estadísticas (fig. 1).

Fig. 1. Estudio de la función hepática por grupos de edades.
A: en la anemia drepanocítica; B: en la hemoglobinopatía SC (B).
En la figura 2 se observa el aumento de la creatinina en la AD con la edad, con diferencias significativas, pero dentro de valores normales (p < 0,05). En la HSC no se presentaron alteraciones.
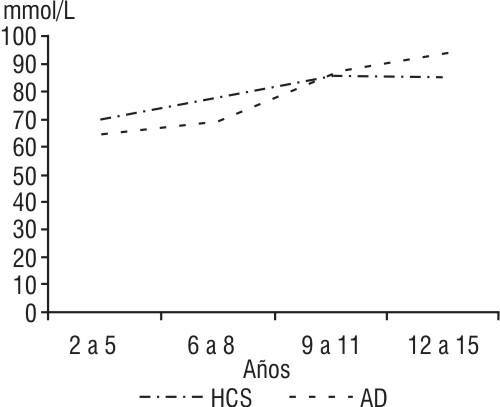
Fig.2. Estudio de la creatinina por grupos de edades en la anemia drepanocítica y en la hemolobinopatía SC.
Discusión
Existen pocos trabajos en los que se estudien parámetros de laboratorio en niños con drepanocitosis.8,13-16
En nuestro estudio los niveles de Hb se encontraron más bajos y los reticulocitos más elevados en la AD que en la HSC, tal como se describe en la literatura. Esto se explica por la mayor intensidad del proceso hemolítico y la expansión del volumen plasmático en la AD.4,17-19 Se comprobó la leucocitosis descrita en otros estudios, que se plantea es la consecuencia de la estimulación de las células progenitoras, de la asplenia y de un recambio de neutrófilos en sangre periférica aumentado.11 Sin embargo, otros autores consideran poco probable que esta alteración sea debida a la autoesplenectomía.20 Los valores de las plaquetas fueron normales, contrario a lo que se describe por otros investigadores,16,21 para lo cual no tenemos una explicación clara.
La ALT, AST y FAS no mostraron alteraciones en ninguna de las hemoglobinopatías, como se describe por otros autores.22,23 La creatinina se mantuvo dentro de valores normales, al igual que lo descrito en otros estudios.18,24 En la HSC fue ligeramente superior al de la AD. Otros autores también encontraron cifras más elevadas de creatinina en la HSC que en la AD, y lo explicaron por la mayor expansión plasmática en esta última.25
La Hb en el sexo femenino fue más elevada con menor reticulocitosis en ambas hemoglobinopatías, dentro de los rangos que se describen en la literatura,26,27 pero solo estadísticamente significativa en la AD. Esto pudiera estar en relación con una Hb fetal (HbF) más elevada en las niñas, ya que un locus ligado al cromosoma X regula la producción de hematíes con HbF (células F).16,28,29 Los leucocitos, plaquetas, ALT, AST, FAS y creatinina, no presentaron diferencias importantes entre los sexos, en ninguna de las hemoglobinopatías.
Se plantea que la Hb desciende rápidamente en los primeros 3 meses de la vida, se mantiene estable de los 3 a 6 meses de edad, disminuye a los 15 meses y no hay cambios relacionados con la edad hasta los 6 años, en que se comienzan a ver variaciones entre los sexos.4,16,28 En nuestro trabajo no encontramos variaciones significativas de los valores de Hb, reticulocitos, leucocitos y plaquetas por grupos de edades, tal como se describe en otras series en niños.18
La ALT y la FAS en la AD mostraron una tendencia ascendente con la edad, con significación estadística, aunque se mantuvieron siempre dentro de rangos normales, lo cual pudiera ser interpretado como signo de lesión hepática que comienza en la infancia, como plantean algunos autores.9-11,22,23,30 La AST no presentó variaciones importantes. El estudio de la creatinina mostró valores bajos, pero dentro de límites normales, igual que en otras series,11,24 pero con un aumento significativo de la creatinina con la edad, que pudiera ser la expresión de un deterioro temprano y progresivo de la función renal, que se hace evidente posteriormente en el adulto.11,21,22 Sin embargo, también se ha descrito aumento de la creatinina en la pubertad por el aumento de la masa muscular. Recientemente se ha planteado que la detección temprana de microalbuminuria y el tratamiento con enalapril, puede retardar la aparición de la insuficiencia renal crónica en el adulto con drepanocitosis.31
Los parámetros hematológicos y de función hepática y renal en la HSC en los diferentes grupos de edades fueron normales, sin diferencias estadísticas.
En general, el cuadro hematológico y el estudio de la función hepática y renal de nuestros pacientes son similares a los descritos en la literatura. El aumento con la edad de algunas pruebas de laboratorio que miden la función hepática y renal, puede ser la expresión de una lesión precoz de esos órganos, cuyo estudio más profundo podría permitir alguna intervención terapéutica para prevenir o detener su evolución.
Summary
A retrospective longitudinal study of some hematological parameters and of hepatic and renal function of 88 children with sickle cell anemia, 35 females and 53 females; 63 with sickle cell anemia and 25 with hemoglobinopathy was conducted from January 1977 to December 1986 in the Hematology and Immunology Institute, with the objective of studying possible disorders and their relation with age, sex and type of hemoglobinopathy. The yearly average rate of hemoglobin, reticulocytes, leukocytes, platelets and the annual values of alanine aminotransferase(ALAT), asparte transferase, serum alkaline phosphatase(SAP) and creatinine were analyzed, grouped in age groups (2-5 y, 6-8 y, 9-11 y and 2-15 y). Hemoglobin and reticulocytes showed significant differences among hemoglobinopathies ( p< 0,05). Hepatic and renal functions were within standard parameters, but it was observed that in sickle cell anemia, ALAT, SAP and creatinine values progressively increases with the age ( p< 0,05). Subject headnigs: ANEMIA, HEMOLYTIC; LONGITUDINAL STUDIES; AGE FACTORS.Referencias Bibliográficas
- Ingram VM. Abonormal human haemoglobin. The comparison of normal human and sickle haemoglobins by fingerprinting. Biochem Biophys Acta 1957; 28: 539.
- Pagnier J, Mears JG, Dunda-Belkodia O, Shafer-Rego KE, Beljord C, Nagel RL, et al. Evidence for the multicentric origen of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci USA 1994; 81: 1771-3.
- El Mouzan MI, Awamy BH, Al Torki MT. Clinal feature of sickle cell disease in Eastern Saudi Arab children. Am J Pediatr Hematol Oncol 1990; 12: 51-5.
- Colombo B, Guerchicoff E, Martínez G. Genética y clínica de las hemoglobinas humanas. La Habana: Editorial Pueblo y Educación; 1993.p. 146-95.
- Rodgers G. Overview of pathophisiology and rational for treatment of sickle cell anemia. Semin Hematol 1997; 34: 2-7.
- Vishinsky ER. Comprehensive care in sickle cell disease. Its impac on morbidity and mortality. Semin Hematol 1981; 28: 220-60.
- Powars D, Chan LS, Schroider WA. The variable expression of sickle cell disease is genetically determined. Semin Hematol 1990; 27: 360-76.
- Platt OS, Gallagher D, Kinny TR, Sloane D, Klug P, Rida W. Mortality in sickle cell disease: Life expectancy and risk factor for early death. N Engl J Med 1994; 330: 1639-44.
- Embury SH, Hebbel RP, Mohandas N, Steinberg MH, eds. Sickle cell disease. Basic principles and clinical practise. New York: Raven; 1994.
- Ataga KI, Orringe EP. Renal abnormalities in sickle cell disease. Am J Hematol 2000; 63: 205-11.
- Wests M, Wethers D, Smith J, Steinberg M. The coopertative study of sickle cell disease. Laboratory profile of sickle cell disease: a cross-selectional analysis. J Clin Epidemol 1992; 45: 893-909.
- Still R, Torrie J. Principles and procedures of statistic. New York: Mc Graw Hill; 1960. p. 406-10.
- Powards DR. Natural history of sickle cell disea: The ten first years. Semin Hematol 1975; 12: 267-85.
- Lee A, Thomas PW, Cupidore L, Serjeant B. Improved survival in homozygous sickle cell disease: lessons from a cohort study. Biomedical J 1995; 311: 1600-2.
- Thomas PW, Higgs DR, Serjeant GR. Benign clinical course in homozygous sickle cell disease: a search for predictors factors. J Clin Epidemiol 1997; 50: 121-6.
- Serjeant GR, Grandinson Y, Lowrie Y, Vaidya S, Mason KP, Phillips J, et al. The development of haematological changes in homozygous sickle cell disease: a cohort study from birth to 6 years. Br J Haematol 1981; 48: 533-43.
- Svarch E, Nordet I, Machín S, Fernández L, Muñiz A, Wade M. La drepanocitosis en los cincos primeros años de la vida. Sangre 1996; 41: 43-6.
- Serjeant GR, Serjeant BE. Sickle cell disease. 3er ed. Oxford: University Press; 2001. p. 107, 111, 115, 126-8, 323.
- Serjeant GR, Serjeant BE, Stephen S. Determinants of haemoglobin level in steady state homozygous sickle cell disease. Br J Haematol 1996; 92: 143-9.
- Boggs DR, Hyde F, Strodes C. An unusual patter of neutrophil kinetic in sickle cell anemia. Blood 1973; 41: 59-65.
- Kenny MW, George AJ, Stuart J. Platelet hyperatictivity in sickle cell disease: a consequence of hyposplenism. J Clin Pathol 1980; 33: 622-5.
- Quintero I, Castañeda C, Villaescusa R, Curbero D, Svarch E. Hepatopatía en niños con anemia drepanocítica. Rev Cubana Pediatr 1984; 56: 25-36.
- Espinosa E, Barreto G, Cabrera J, Hernández P. El hígado en la anemia drepanocítica. Un estudio clínico patológico en 50 pacientes. Sangre 1985; 30: 945.
- Al-Noama LM, al-Sadoon EA, al-Sadoon TA. Levels of uric acid, urea and creatinine in Iraqui children with sickle cell disease. JPMA J Pak Med Assoc 2000; 50: 98-102.
- Ballas SK, Lewis CN, Noone AM, Krasnow SH, Kamarulzaman E, Burka ER. Clinical hematological and biochemical features of HSC disease. Am J Hematol 1982; 13: 37-51.
- Morris J, Dunn D, Bickford M. The haematology of homozygous sickle cell disease. Br J Haematol 1991; 77: 382-5.
- García T, Nordet I, Machín S, González A, Muñiz A, Martínez G, et al. Aporte al estudio de la drepanocitosis. Análisis clínico y hematológico en los primeros 5 años de la vida. Rev Cubana Hematol Inmunol Hemoter 1999; 15: 96-104.
- Chang YC, Maier-Redelsperger M, Smith KD, Contul DR, De Montalembert M. The relative importance of X-linked FCP locus and b-globin haplotypes in detemining haemoglobin F leves: a study of SS patients homozygous for S haplotypes. Br J Haematol 1997; 96: 806-14.
- Hayes RJ, Beckford M, Grandinson Y, Mason K, Serjeant GR. The haematology of the steady state homozygous sickle cell disease: frequency distributions, variations with age and sex, longitudinal observation. Br J Haematol 1985; 59: 369-82.
- Svarch E, Espinosa E, Hernández P, Martínez G, Ballester J. Resultados de los estudios realizados en Cuba sobre hemoglobinopatías S. Sangre 1991; 36: 37-42.
- Foucan L, Bourhis V, Bangou J, Marault L, Etienne-Julian M, Salmi RL. A randomized trial of captopril for microalbuminuria in normotensive adults with sickle cell anemia. Am J Med 1998; 194: 339-42.
Recibido: 23 de febrero de 2003. Aprobado:10 de marzo del 2003.
Dr. Sergio A. Machín García. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Cuidad de La Habana, Cuba. Tel (537)578268. Fax (537) 442334.e-mail: ihidir@hemato.sld.cu
1Instituto de Hematología e Inmunología.
2Dirección de Servicios Médicos Internacionales.

