










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.26 n.1 Ciudad de la Habana ene.-mar. 2010
PRESENTACIÓN DE CASOS
Enfermedad de Kikuchi-Fujimoto. Presentación de un caso pediátrico
Kikuchi-Fujimoto disease: Presentation of a pediatric case
Dra. Ania Hernández CabezasI; Dra. Nelkis C. Díaz LinaresII
IInstituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
IIHospital Pediátrico "William Soler". Ciudad de La Habana, Cuba.
RESUMEN
Se comunica un paciente masculino de 14 años, blanco, con diagnóstico de enfermedad de Kikuchi-Fujimoto, que presentaba adenopatías predominantemente en región cervical izquierda, fiebre de corto tiempo de evolución y compromiso del estado general sin pérdida de peso. Se hicieron estudios de hematología general, serología viral y biopsia por aspiración con aguja fina de un ganglio cervical. El aspirado fue sospechoso de células neoplásicas. La biopsia ganglionar mostró abundantes histiocitos, inmunoblasblos, necrosis celular con polvo nuclear y cariorrexis, así como ausencia de neutrófilos y eosinófilos. Evolucionó favorablemente con desaparición de la fiebre a los 20 días del inicio de sus manifestaciones con regresión paulatina de las adenopatías.
Palabras claves: enfermedad de Kikuchi-Fujimoto, adenopatías, fiebre.
ABSTRACT
This is the case of a white patient aged 14 diagnosed with Kikuchi-Fujimoto disease presenting adenopathies prevailing in left cervical region, short-term course fever and involvement of its general condition without weigh loss. Authors made studies of general hematology, viral serology and fine needle aspiration biopsy of a cervical ganglion. There were suspicions of neoplasic cells in aspirate. Ganglion biopsy showed abundant histiocytes, immunoblasts, and cellular necrosis with nuclear powder and caryorrhesis, as well as a lack of neutrophils and eosinophils. There was a favorable course with disappearance of fever at 20 days from the onset of its manifestations with a gradual regression of adenopathies.
Key words: Kikuchi-Fujimoto disease, adenopathies, fever.
INTRODUCCIÓN
La enfermedad de Kikuchi-Fujimoto (KF) es un proceso benigno de causa desconocida, al que se han encontrado asociadas enfermedades autoinmunes1 o virales. Se conoce también como linfadenitis histiocítica necrotizante, y está caracterizada por afectación ganglionar con mayor frecuencia cervical unilateral, algunas veces bilateral, aunque esta puede ser sistémica e indolora, con predominio en el sexo femenino, especialmente en menores de 30 años.1-4 La presentación en niños no es frecuente, aunque se ha descrito en ocasiones.5-7 La linfoadenopatía de KF puede acompañarse por otros síntomas: fiebre moderada, mialgias, erupción cutánea y dolor localizado.2,8 En algunos casos puede haber leucopenia con linfocitosis moderada.9 Esta entidad se describió por primera vez en Asia en el año 1972 y resuelve en 2 ó 3 meses, aunque puede haber recurrencia a largo plazo.7
PRESENTACIÓN DEL CASO
Paciente masculino, blanco, de 14 años de edad, con antecedentes de buena salud, que comenzó con fiebre después de haberse bañado en una piscina. A los 5 días de la fiebre notó aumento de volumen de ganglios cervicales bilaterales con predominio de la cadena lateral izquierda. Presentaba una gran astenia.
Al examen físico se comprobaron las adenopatías referidas que eran de consistencia blanda fusionadas en forma de un paquete multinodular, indoloro. La dimensión del ganglio mayor era de 2 × 1,5 cm. Existían otras adenomegalias más pequeñas y con similares características, en la región maxilar izquierda. No se encontraron adenopatías inguinales ni existía hepato o esplenomegalia.
Los exámenes realizados revelaron: Hb 14 g/dL, leucocitos 8,5 × 109/L, linfocitos 51, segmentados 39, eosinófilos 6, monocitos 4, plaquetas 300 × 109/L y eritrosedimentación 38 mm/h. Los estudios bioquímicos indicados fueron normales, así como el ultrasonido abdominal, la radiografía de tórax y la TAC de tórax y abdomen.
Los estudios serológicos para el virus de Epstein Barr, citomegalovirus y toxoplasma fueron todos igualmente negativos.
Se le indicó tratamiento empírico con antibióticos y biopsia de un ganglio cervical por aspiración con aguja fina (BAAF).
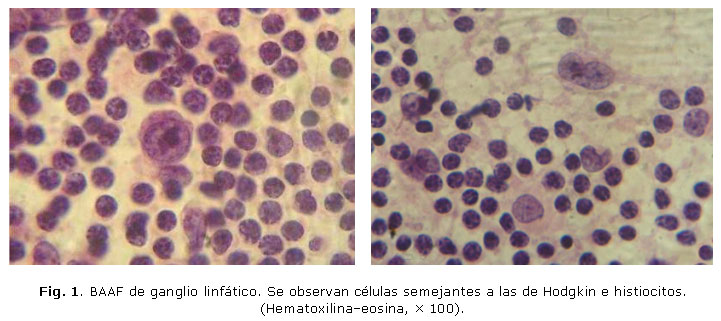
En el extendido del aspirado ganglionar se observaron células de aspecto histiocitario y otras con nucleolo prominente, que sugerían células de aspecto semejante a las de Hodgkin (Fig. 1). Había escasas células plasmáticas y linfocitos.
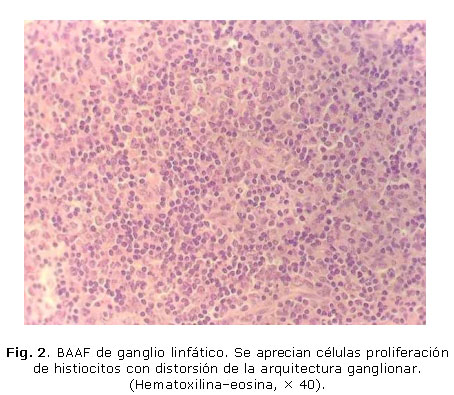
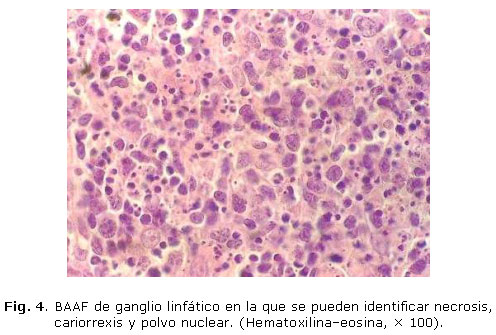
La biopsia ganglionar mostró una arquitectura distorsionada con folículos residuales y una proliferación de histiocitos prominentes (Fig. 2). Además, se observaron linfocitos, ausencia de neutrófilos y focos de necrosis a predominio de células aisladas con cariorrexis y polvo nuclear (Figs. 3 y 4).

El estudio inmunoquímico de la preparación ganglionar ofreció los siguientes resultados: CD20 (±), CD3 (+), CD68 (+), CD30 (-). En general, el aspecto histológico resultó compatible con una linfadenitis necrotizante de Kikuchi-Fujimoto.
Evolutivamente, el paciente mejoró su estado general, la fiebre desapareció a los 7 días después de practicada la biopsia y teniendo en cuenta la asociación de esta enfermedad con algunos procesos autoinmunes y virales, se completaron las investigaciones con la determinación de anticuerpos antinucleares y factor reumatoideo, que fueron negativos. El paciente fue dado de alta con un seguimiento evolutivo satisfactorio.
DISCUSIÓN
La enfermedad de KF no se manifiesta con frecuencia en la edad pediátrica. La presencia de histiocitos y células mononucleares de aspecto inmunoblástico con características semejantes a las de Hodgkin, con abundantes linfocitos y algunas células plasmáticas, inclinaron hacia la posibilidad de un proceso linfoproliferativo y fue el estudio histológico el que permitió esclarecer el diagnóstico.
Esta enfermedad se describió en 1972 por Kikuchi10 y Fujimoto11 como una linfadenitis necrotizante que afectaba predominantemente a mujeres de origen asiático y menores de 30 años, aunque hay reportes en que aproximadamente el 10 % se corresponde con pacientes de alrededor de 20 años. La relación mujer/hombre es 4:1.
La presentación clínica habitual es una adenopatía cervical, única, bilateral cervical o múltiple, y fiebre. Entre el 40 y 50 % de los casos presentan fatiga, dolores osteomusculares, pérdida de peso y sudoraciones nocturnas. En el 30 % de los enfermos se pudieran ver manifestaciones cutáneas, ya sea mácula, pápula o eritema urticariano. La hepatoesplenomegalia se ha comunicado en el 10 % de los casos y más raramente se puede encontrar compromiso neurológico, meningitis aséptica o ataxia cerebelosa.12
En otros pacientes se ha manifestado como un síndrome febril prolongado de etiología no precisada y además, en ocasiones se han descrito recurrencias a largo plazo, como parte de un síndrome febril o adenomegálico.7
Los estudios serológicos no han demostrado un agente etiológico infeccioso, aunque se ha visto asociación con el virus de Epstein-Barr, la Yersinia enterocolítica, el parvovirus, la Brucella, la Bastonella henselae, el citomegalovirus y la toxoplasmosis.11,13-15
En los estudios de laboratorio no se han comunicado cambios específicos, solo aumento de la eritrosedimentación,14 tal como sucedió en nuestro paciente.
En el hemograma puede haber neutropenia, linfocitosis con linfocitos atípicos y en algunos casos puede existir alteración de la función hepática, así como deshidrogenasa láctica elevada.
La patogénesis de esta enfermedad no es totalmente conocida. Algunos autores plantean que se trata de una enfermedad autoinmune similar al lupus eritematoso, desencadenada por un virus que infecta y transforma a los linfocitos, o por una reacción hiperinmune de células T e histiocitos activados por un patógeno no identificado que induce una degeneración celular y necrosis ganglionar. Esto estaría apoyado por el hecho de que se han descrito pacientes que desarrollan enfermedades del tejido conectivo a largo plazo después de padecer una enfermedad de KF.10
Se ha visto también en esta entidad un cambio en la relación de los linfocitos T CD4/CD8 después del primer mes de evolución, con predominio inicial de CD8, para más tarde predominar los CD4.3
Por otra parte, hay autores que sugieren que no hay una causa única de enfermedad de KF, sino que es el resultado de una estimulación inmunológica de varios agentes infecciosos y estímulos ambientales.3
La histopatología del ganglio en la enfermedad de KF aporta los elementos para su diagnóstico y estos incluyen una arquitectura ganglionar parcialmente mantenida con folículos residuales que muestran hiperplasia de centros germinales reactivos, áreas en parches de necrosis comúnmente en la región cortical o paracortical.
Algunas veces, el tejido necrótico es solamente focal o de células individuales y en estos casos, se ven agregados de histiocitos con detritus celulares o polvo nuclear.
Las áreas de proliferación de histiocitos están acompañadas de linfocitos, linfocitos activados y moderada cantidad de células plasmáticas; los eosinófilos y los neutrófilos están ausentes de manera distintiva en esta entidad. En la periferia de la lesión se aprecian nidos de células T plasmocitoides, linfocitos grandes con células plasmáticas y se identifican marcadores de células T.
Algunos autores consideran la proliferación histiocítica, más que la necrosis, el cuadro característico de la linfadenitis de KF y aún incluyen en esta entidad pacientes con ausencia de necrosis que muestran solamente escasas células con picnosis y cariorrexis, y un número moderado de mitosis.
En dependencia del cuadro histológico, se distinguen 4 subtipos: linfohistiocítico, histiocítico con necrosis de células aisladas, necrótico y de células espumosas.8 El diagnóstico diferencial incluye las linfadenitis necrotizantes, ya sean infecciosas como la tuberculosis, histoplasmosis, enfermedad por arañazo de gato, sífilis, reacciones alérgicas, autoinmunes como el lupus eritematoso y las malignas como el linfoma de Hodgkin o los no hodgkinianos.
En nuestro caso, se descartaron sin dificultad un número amplio de estas enfermedades, pues no había necrosis caseosa, vasculitis ni alteraciones clínicas e histológicas compatibles con los procesos. Sin embargo, era necesario establecer el diagnóstico diferencial con el linfoma de Hodgkin, sobre todo atendiendo al estudio citológico, teniendo en consideración la presencia de abundantes histiocitos, linfocitos, algunas células plasmáticas e inmunoblastos con aspecto similar a las células del tipo Hodgkin. Esta situación fue esclarecida por la biopsia.
En la literatura se ha indicado que de 108 biopsias practicadas en pacientes con enfermedad de KF, el 30 % fue mal diagnosticada como linfoma, fundamentalmente por la proliferación de células histiocíticas e inmunoblastos que no se correspondían con una proliferación monoclonal de linfocitos.16,17
El tratamiento es inespecífico y se han utilizado antibióticos, antiinflamatorios no esteroideos y corticoides; estos últimos en casos de hiperpirexias y compromiso meníngeo o neurológico severo. La resolución espontánea de los síntomas es la regla, y ocurre entre 1 a 4 meses después de iniciada la enfermedad.14 En una serie publicada de 64 enfermos, 59 ya estaban bien a los 32 meses de seguimiento.16 En los casos que han fallecido, la mayoría ha sido como consecuencia de un compromiso cardíaco o meníngeo.
Nuestro trabajo aporta un nuevo caso pediátrico que confirma que aunque la enfermedad de KF es rara en niños, también en ellos es necesario tener en cuenta esta posibilidad diagnóstica.
REFERENCIAS BIBLIOGRÁFICAS
1. Norris AH, Krasinskas AM, Salhany K. Kikuchi-Fujimoto disease: A benign cause of fever and lymphadenopathy. Am J Med 1996;171:401-5.
2. Imamura M, Ueno H, Matsuura A, Kamiya H, Suzuki T, Kikuchi K, et al. An ultraestructural study of subacute necrotizing lymphadenitis. Am J Pathol 1982;107:292-9.
3. Etcheverry R, Armas Cruz R, Martínez V. Linfoadenitis necrotizante subaguda (Enfermedad de Kikuchi y Fujimoto). Rev Med Chile 1990;118:431-6.
4. Kucukardali Y, Solmazqul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi- Fujimoto disease: Analisis of 244 cases. Clin Rheumatol 2007;26:50-4.
5. Adjaoud D, Boudjemaa S, Boccon-Gibod L, Leverger G. Kikuchi- Fujimoto's disease: Report of 5 cases and literature review. Arch Pediatr 2007;14:1333-6.
6. Chase SP, Templer J W, Miick R, Diaz- Arias AA. Cervical lymphadenopathy secondary to Kikuchi-Fujimoto disease in child: Case report. Ear Nose Throat J 2008;87:350-3.
7. Dylewski J, Berry G,Pham-Dang H. An unusual cause of cervical lymphadenitis: Kikuchi-Fujimoto disease. Rev Infect Dis 1991;13:823-5.
8. Tordecilla C Juan, Medina M, Ávila R, Campbell M. Enfermedad de Kikuchi Fujimoto. Rev Chil Pediatr 2002;73:483-8.
9. Rohmann I, Bentjerody R. Linfoadenitis necrotizante de Kikuchi: Histopatología en 2 casos. Rev Med Chile 1988;116:929-33.
10. Fujimoto Y, Kosima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis. A new clinicopathologic agent. Naika 1972;20:920-7.
11. Dorfman RF, Berry GJ. Kikuchi¨s histiocytic necrotizing lymphadenitis: An analisis of 108 cases with emphasis on differential diagnosis. Sem Diagn Pathol 1988;5:329-45.
12. Debley J, Rozansky D, Miller M, Katz B, Greene, M. Histiocytic necrotizing lymphadenitis with autoinmune phenomena and meningitis in a 14 year-old girl. Pediatrics 1996;98:130-3.
13. Rodríguez J, Martín M, Báez J, Gil J. Kikuchi_Fujimoto necrotizing lymphadenitis associated with brucellosis. Sangre 1992;37:201-4.
14. Murga-Sierra M, Vegas E, Blanco-González J, González A, Martínez P, Calero M. Kikuchi's disease with multisystemic involvement and adverse reaction to drugs. Pediatrics 1999;104:e24.
15. Stephan JL, Jeannoel P, Chanoz J, Gentil-Perret A.Epstein-Barr virus-associated Kikuchi disease in two children. J Ped Hematol Oncol 2001;23:240-3.
16. Seo JH, Shim HS, Park JJ, Jeon SY, K Wong OJ. A clinical study of histiocytic necrotizing lymphadenitis (Kikuchi's disease) in children. Int J Pediatr Otorhinolaryngol 2008;72:1637-42.
17. Kim CH, Hyun OJ, Yoo Jer, Kim SH, Sohn HS, Chung SK. Kikuchi disease mimicking malignant lymphoma on PDG PET/Ct. Clin Nucl Med 2007;32:711-2.
Recibido: 10 de noviembre de 2009.
Aprobado: 25 de noviembre de 2009.
Dra. Ania Hernández Cabezas. Instituto de Hematología e Inmunología. Apartado 8070. Ciudad de La Habana, Cuba, CP 10800, Cuba. Tel (537) 643 8695 8268, Fax (537) 644 2334. E-mail: ihidir@hemato.sld.cu y http://www.sld.cu/sitios/ihi


{kind=link}