










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.26 n.3 Ciudad de la Habana sep.-dic. 2010
COMUNICACIÓN BREVE
Compound heterozygosity of Hb QIndia (a64 (E13) ASP®HIS) and -a3,7 thalassemia. First report from Argentina
Heterocigosis de la Hb QIndia (a64 (E13) ASP ®HIS) y -a3,7 thalassemia. Primer informe desde Argentina
Dr. Susana M. PérezI; Dr. Nélida I. NogueraI; Dr. Irma del Luján AcostaI; Dr. Karina Lucrecia CalvoI; Dr. Irma Margarita BragósI; Dr. Virginia ResciaII; Dr. Ángela MilaniI
IDepartamento de Bioquímica Clínica, Cátedra de Hematología. Facultad de Ciencias Bioquímicas y Farmacéuticas. Universidad Nacional de Rosario. Rosario, Argentina.
IIServicio de Hematología. Fundación de la Hemofilia. Sala IX, Hospital Provincial del Centenario. Rosario. Argentina.
ABSTRACT
Hemoglobine (Hb) Q-India is an innocuous aglobin variant: a64 Asp ® His. DNA sequencing studies have shown that the Hb Q India mutation is GAC ® CAC in codon 64 of the a1 gene. Hb Q-India is a well-known hemoglobin variant in South-East Asia but only isolated case reports exist in literature to describe this rare entity in the rest of de world. The variant has been found with various forms of aand ß thalassemia. This hemoglobin has the same electrophoretic mobility as Hb S. We report, for the first time, the identification of Hb Q-India in an Argentinian woman (her parents came from Gibraltar), referred to our laboratory bearing a mild microcytic hypocromic anemia; a co-inherited a+ thalassemia (-a3.7 th) was also found.
Key words: Abnormal hemoglobin (Hb), microcytic hypocromic anemia, Hb Q India.
RESUMEN
La hemoglobina (Hb) Q India es una hemoglobina anormal e inocua que afecta la cadena a de esta. Los análisis de secuencia han demostrado que la mutación se encuentra en el codon 64 GAC ® CAC del gen a1. Si bien es una variante muy conocida en el sudeste asiático, solo se han reportado pocos casos en el resto del mundo. Esta hemoglobina anormal se ha encontrado asociada con diversas formas de a y ß talasemia y su posición electroforética es idéntica a la de la Hb S. Reportamos, por primera vez, la identificación de la Hb Q India en una mujer Argentina (cuyos padres procedían del Peñón de Gibraltar), enviada a nuestro laboratorio por padecer de anemia microcítica hipocrómica, en la que se encontró también la coexistencia de a+ talasemia (-a3,7 th).
Palabras clave: hemoglobinas anormales (Hb), anemia microcítica hipocrómica, Hb Q India.
Hemoglobin (Hb) Q disorders are an important group of hemoglobinopathies. Several variants including Hb Q Thailand, Hb Q Iran and Hb Q India are documented.1 Only isolated case reports exist in literature to describe this rare entity.2 They occur normally in the heterozygous form and could be associated with thalassemia.3-7 The three Hb Q variants do not cause hematological disorders because the residues involved are on the surface of the hemoglobin tetramer and charge changes at these positions do not affect the properties of the hemoglobin molecule. This particular Hb Q variant, Hb Q-India, is an a chain variant: a64 Asp ® His. DNA sequencing studies have shown that the Hb Q India mutation is GAC ® CAC in codon 64 of the a1 gene.8,9 We hereby report, for the first time, the identification of Hb Q-India in an Argentinian women, in association with a+ thalassemia (-a3.7 deletion), referred to our laboratory bearing a mild microcytic hypocromic anemia.
Hematological data were obtained with a Coulter Counter model ACT10 (Coulter Corporation, USA). Hb A2 was measured by elution post electrophoresis at alkaline pH, 10 and Hb F according to the method described by Betke et al.10
Isopropanol (Carrell & Kay)11 and heat stability tests were performed. Cellulose acetate electrophoresis at alkaline pH; citrate agar electrophoresis at pH 6; and electrophoresis at alkaline pH of globin chains, were carried out using standard methods.
Sickling test was negative. The isopropanol an the heat test were also negative indicating the absence of an unstable Hb.
Cellulose acetate (alkaline pH) electrophoresis detected Hb X moving to Hb S position and citrate agar (acid pH), moving Hb X as a sharp band close to Hb A. Globin chain electrophoresis enables the identification of an alpha chain alteration.
The hematological data of the patient are shown in table.

DNA was extracted from peripheral blood cells by standard methods.12
The sample was analyzed by allele specific amplification of -a3.7 deletion. 13 Selective amplification of the a2-globin gene was performed in a thermal mini cycler (MJ Research, Watertown, MA). Amplification was accomplished according to conditions already described.13
Polymerase chain reaction (PCR) was carried out and then a direct DNA sequencing of the PCR products was performed. PCR amplification of the a2- and a1-globin genes was accomplished by using oligonucleotide primers (CyberSyn, Lenni, PA, USA); the common forward primers FAa2:
5'-CGCGCTCGCGGCCCGGCAC-3',
and reverse specific primers for the a2 gene:
5'-GGGAGGCCCATCGGGCAGGAGGAAC-3'
and a1 gene: 5'-GGGGGGAGGCCCAAGGGGCAAGAA-3'.
Reverse primers and reaction conditions used are the ones described to detect a-thal-2 (-3.7 kb).14 Sequencing was done using a Big Dye Terminators Ready Reaction Kit (Perkin-Elmer Cetus, Norwalk, CT, USA) in an ABI PRISM 310 sequencer (Perkin-Elmer Cetus). Primers used for sequencing the two genes were the following: exon 1, common forward primers FAa2; exon 2, primer S2
(5'-CCCGCCCGGACCCACA-3'); exon 3, primer S3
(5'-GCGGGTTGCGGGAGGT-3').15 The reverse specific primers for the a1 gene were used to confirm the mutation (fig.).
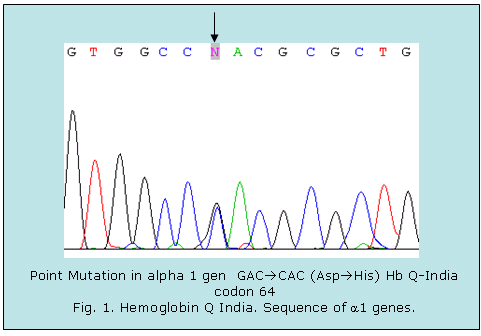
Molecular biology studies (PCR) showed the presence of -a3,7 deletion, and the sequencing of a-1 gene showed a GAC ® CAC (Asp ® His) substitution at codon 64, corresponding to Hb Q India (fig.). Even though Hb Q India is detectable by electrophoresis its correct characterization requires high-performance liquid chromatography (HPLC) or molecular analysis.
It is the first time that this abnormal hemoglobin is described in our country. The low red blood cell indexes observed in this case could be due to co-inheritance of a+ thalassaemia (-a3.7 deletion).
REFERENCES
1. Lorkin PA, Charlesworth D, Lehman H, Rahbar S, Tuchinda LI, Lie I, et al. Two haemoglobins Q, alpha-74 (EF3) and alpha-75 (EF4) aspartic acid to histidine. Br J Haematol 1970;19:117-25.
2. Desai DV, Dhanani H, Kapoor AK, Yeluri SV. HbQ-India in a Sindhi family: An uncommon hemoglobin variant. [Case Reports]. J Lab Hematol 2004;10:212-4.
3. Sukumaran PK, Merchant SM, Desai MP, Lehmann H. Hemoglobin Q India (alpha 64(E13) aspartic acid to histidine) associated with beta-thalassemia observed in three Sindhi families. J Med Genet 1972;9:436-42.
4. Dash S, Huisman TH. Hemoglobin-Q-India (64 (E13) Asp-His) and beta thalassemia: A case report from Punjab (North India). Eur J Haematol 1988;40:281-3.
5. Schmidt RM, Bechtel KC, Moo-Penn WF. Hemoglobin Q India, alpha 64 (E13) Asp replaced by His, and beta-thalassemia in a Canadian family. Am J Clin Pathol 1976;66:446-8.
6. Sagnet H, Morineaud JP, Delprat J, Revil H, Thomas J, Philibert E, et al. A rare hemoglobinosis: The association of hemoglobin Q and alpha-thalassemia. Med Trop 1968;28:133-8.
7. Viprakasit V, Chinchang W, Pung-Amritt P, Tanphaichitr VS. Identification of Hb Q-India (alpha64 Asp—>His) in Thailand. Hematology 2004;9:151-5.
8. Abraham R, Thomas M, Britt R, Fisher C, Old J. Hb Q-India: An uncommon variant diagnosed in three Punjabi patients with diabetes is identified by a novel DNA analysis test. J Clin Pathol 2003;56:296-9.
9. Chenna R, Sugawara H, Koike T, López R, Gibson T, Higgins D, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 2003;31:3497-500.
10. Efremov GD, Huisman THF. Diagnóstico de laboratorio de Hemoglobinopatías en Hemoglobinas anormales. En: Weatherall DJ. Clínica Hematológica 2/2. Barcelona: Salvat; 1976. p. A) 319-320 B) 322-323.
11. Carrell RW, Kay R. A simple method for the detection of unstable haemoglobins. Br J Haematol 1972;23:615-9.
12. Miller SA, Dykes DD, Polesky HF. A simple salting-out procedure for extracting DNA from human nucleated cells. Nuclei Acids Res 1988;16:1215.
13. Smetanina NS, Huisman THJ. Detection of α-thalassemia-2 (-3.7 kb) and its corresponding triplication aaa (anti 3.7 kb) by PCR; an improved technical change. Am J Hematol 1996;53,201-3.
14. Molchanova, TP, Pobedimskaya DD, Postnikov VA. A simplified procedure for sequencing amplified DNA containing the α2- or α1-globin gene. Hemoglobin 1994;18:251-5.
15. Noguera NI, González FA, Dávoli RA, Milani AC, Villegas A. A novel splices acceptor site mutation of the alpha2-globin gene causing alpha-thalassemia. Hemoglobin 2001;25:311-5.
Recibido: 24 de marzo del 2010.
Aprobado: 12 de abril del 2010.
Dra. Susana M. Pérez. Departamento de Bioquímica Clínica, Cátedra de Hematología. Facultad de Ciencias Bioquímicas y Farmacéuticas. Universidad Nacional de Rosario. Rosario, Argentina. e-mail: sperez@fbioyf.unr.edu.ar
Corresponding author: +54-341-4804598; e-mail: sperez@fbioyf.unr.edu.ar

