










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.27 n.1 Ciudad de la Habana ene.-mar. 2011
ARTÍCULOS ORIGINALES
La drepanocitosis en Cuba. Estudio en niños
Drepanocytosis in Cuba: Study in children
Prof. DraC. Eva Svarch,I DraC. Beatriz Marcheco-Teruel,II Dr. Sergio Machín-García,I Dra. Andrea Menéndez-Veitía,I Dra. Ileana Nordet-Carrera,I Dr. Alberto Arencibia-Núñez,I Dr. Aramís Núñez-Quintana,III Lic. Raúl Martínez-Triana,I Dr. Claudio Scherle-Matamoros,III Dr. Eliécer San Román-García,IV Dr. Alejandro González-Otero,I Prof. DrC. Ernesto de la Torre-MontejoI
I Instituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
II Centro Nacional de Genética Médica. Ciudad de La Habana, Cuba.
III Hospital Clinicoquirúrgico "Hermanos Ameijeiras". Ciudad de La Habana, Cuba.
IV Instituto de Cardiología y Cirugía Cardiovascular. Ciudad de La Habana, Cuba.
RESUMEN
La drepanocitosis es la anemia hemolítica determinada genéticamente más frecuente en el mundo. En Cuba, la frecuencia del estado de portador es del 3,08 % en la población general. La fisiopatología de la oclusión vascular es muy compleja; involucra la polimerización de la Hb S, las alteraciones de la membrana del hematíe, las moléculas de adhesión, las citocinas inflamatorias, los factores de la coagulación y lesiones del endotelio vascular. Las manifestaciones clínicas más frecuentes son: las crisis vasooclusivas dolorosas, el síndrome torácico agudo, la crisis de secuestro esplénico, la crisis aplástica, la necrosis aséptica de la cabeza del fémur y la úlcera maleolar. El cuadro clínico es muy variable: desde niños que mueren temprano en la vida hasta pacientes que alcanzan la sexta década de la vida. En el Instituto de Hematología e Inmunología existe un Programa de Atención Integral que incluye: seguimiento sistemático desde temprano en la vida en una consulta especializada, la administración de ácido fólico de forma permanente y de penicilina oral profiláctica los primeros 5 años de la vida; así como la educación del niño y de sus padres. Desde 1986 se realiza esplenectomía parcial en la crisis de secuestro esplénico con excelentes resultados. Entre 2004-2008 fallecieron solamente 16 enfermos en todo el país y en 397 adultos la sobrevida fue de 53 años en la anemia drepanocítica y de 58 en la hemoglobinopatía SC. Como resultado de este programa, en los últimos años la sobrevida ha aumentado, la calidad de vida del paciente ha mejorado y han disminuido los costos invertidos en el tratamiento de las complicaciones.
Palabras clave: drepanocitosis, crisis vasooclusivas, síndrome torácico, crisis de secuestro esplénico, esplenectomía parcial, hidroxiurea.
ABSTRACT
Drepanocytosis is the hemolytic anemia more frequent genetically determined in the world. In Cuba, the frequency of carrier status is of 3,08% in general population. The pathophysiology of vascular occlusion is very complex; includes the polymerization of the Hb S, the alterations of red-blood cells, the adhesion molecules, the inflammatory cytokines, the coagulation factors and the lesions of the vascular endothelium. The more frequent clinical manifestations are: painful vaso-occlusive crises, the acute thoracic syndrome, the splenic sequestration crisis, the aplastic crisis, the aseptic necrosis of femur head and malleolar ulcer. The clinical picture is very variable: from children dying early in life up to patients achieve the sixth decade of life. In the Institute of Hematology and Immunology there is an Integral Care Program including: systematic follow-up from early in life in a specialized consultation, permanent administration of folic acid and of prophylactic oral penicillin during the first 5 years of age; as well as the child education and of parents. From 1986 it is carried out the partial splenectomy in crises of splenic sequestration with excellent results. Between 2004-2008 in all the country deceased only 16 patients and in 397 adults the survival rate was of 53 years in the drepanocythemia and of 58 in the SC hemoglobinopathy. As result of this program, in past years the survival has increased, the quality of life of patient improved and the costs spent in treatment of complications has decrease.
Key words: Drepanocytosis, vaso-occlusive crisis, thoracic syndrome, splenic sequestration crisis, partial splenectomy, hydroxyurea.
INTRODUCCIÓN
La drepanocitosis es la anemia hemolítica determinada genéticamente más frecuente en el mundo. Se debe a una mutación en la posición 6 del gen que codifica para la cadena b de la globina en la que el ácido glutámico es remplazado por la valina. Su frecuencia es mayor en África, el Medio Oriente, sur de Italia, norte de Grecia, sur de Turquía, provincias occidentales de Arabia Saudita y la India. Fue trasladada por el comercio de esclavos a Estados Unidos de Norteamérica, América Central, el Caribe y algunos países de América del Sur.
En el mundo la frecuencia del estado de portador AS es del 7 % y cada año nacen de 300 000 - 400 000 niños enfermos.1
Es la hemoglobinopatía más frecuente en los EE. UU. y en muchos países subdesarrollados, pero también se considera un problema de salud en Europa, debido a que la inmigración desde países en los que la enfermedad es prevalente, aumentó de manera considerable en los últimos 15 años. En la actualidad, se calcula que existen en ese continente 1 500 000 portadores AS.2 En muchos países de América Latina y del Caribe ha cobrado más importancia en los últimos años debido a que se ha logrado una disminución de la mortalidad por enfermedades respiratorias, diarreicas agudas y desnutrición.3
La forma más frecuente de drepanocitosis en Cuba es la anemia drepanocítica (AD) o hemoglobinopatía SS. Le siguen en frecuencia: la hemoglobinopatía SC (HSC) y la Sb talasemia (Sb tal); esta última puede ser Sb0 tal o Sb+tal.4
FISIOPATOLOGÍA
El evento primario de la fisiopatología es la polimerización de la hemoglobina (Hb) S desoxigenada que deforma al hematíe. Este hematíe deformado (drepanocito), ocluye frecuentemente la microcirculación, lo que provoca hipoxia, más polimerización y por consiguiente, más oclusión vascular. Este proceso aparentemente simple, es en realidad muy complejo; en él participa una disminución del óxido nítrico debido a su captación por la Hb liberada en el plasma por la hemólisis intravascular. Esta disminución produce vasoconstricción, aumento de la expresión de moléculas de adhesión y activación plaquetaria. Existen alteraciones de la membrana del hematíe que conducen a una homeostasis anormal de cationes con deshidratación de la célula y la expresión de fosfatidilserina (procoagulante) en su superficie. Participan en el proceso: citocinas inflamatorias, factores de la coagulación, una disminución de la enzima Adams 13 con aumento de los multímeros del factor von Willebrand, y alteraciones reológicas y hemodinámicas. La microvasculatura tiene un tono anormal y se produce una vasculopatía proliferativa por aumento del factor inducido por la hipoxia (HIF-1). El glicocalix, sustancia antiinflamatoria y antiadhesiva que protege al endotelio vascular de su contacto con las células sanguíneas circulantes, disminuye por hipoxia e isquemia, y se produce un aumento del factor de necrosis tumoral (FNT a) que lesiona el endotelio vascular.
La oclusión vascular es considerada en la actualidad, una forma de injuria de repercusión, en la que el estrés oxidativo y la inflamación llevan al daño crónico de los órganos.5-8
CUADRO CLÍNICO
Las manifestaciones clínicas más frecuentes y que en gran medida determinan la severidad del cuadro clínico, son las crisis vasooclusivas dolorosas (CVOD) y el síndrome torácico agudo (STA). Las CVOD se definen como dolor de más de 4 horas de duración, en cualquier sitio del organismo. Una forma particular de CVOD es la dactilitis que se presenta en el niño menor de 4 años con dolor y signos inflamatorios en los dedos de las manos o pies o en ambos, y que es casi patognomónica.4
El STA se define como la aparición de nuevas lesiones inflamatorias en la radiografía de tórax que casi siempre se acompañan de fiebre y manifestaciones respiratorias. Su etiología más frecuente es la infección por neumococo, hemofilus, micoplasma, clamidia y virus, pero también se produce por infarto o tromboembolismo y con frecuencia, por una combinación de estos procesos.9 El STA es la causa más frecuente de hospitalización y muerte en la drepanocitosis.10 El tratamiento consiste en la administración rápida de antibióticos que actúen sobre los gérmenes más comunes en el proceso. Si el cuadro es severo y la Hb se encuentra por debajo de 8g/L, se debe indicar transfusión de glóbulos rojos, que no solo mejora la oxigenación, sino que neutraliza los ácidos grasos libres que lesionan el pulmón.9 Se plantea que un polimorfismo en los genes de la endotelina 1 y de la óxido nítrico sintetasa se relacionan con la frecuencia del STA.10
En el niño con drepanocitosis, las infecciones son 400 veces más frecuentes que en el niño normal, sobre todo por neumococos.8 La causa más importante de esta susceptibilidad es la asplenia funcional, aunque existen otras alteraciones inmunológicas.
Otras manifestaciones son: la crisis aguda de secuestro esplénico, la crisis aplástica, el accidente vascular encefálico (AVE) y la necrosis aséptica de la cabeza del fémur.
La úlcera maleolar y la litiasis vesicular son poco frecuentes en el niño.
En la actualidad, se definen 2 subfenotipos: uno determinado fundamentalmente por la oclusión vascular y caracterizado por CVOD, STA y osteonecrosis; y otro que depende de la hemólisis intravascular con disminución del óxido nítrico y aumento de la adhesión y agregación plaquetaria, que incluye: hipertensión pulmonar, priapismo, úlcera maleolar y AVE.11
La oclusión vascular, constante y subclínica, lleva al daño crónico de los órganos: insuficiencia renal crónica, alteraciones hepáticas, pulmonares, cardiovasculares, óseas y oftalmológicas.3,4
VARIABILIDAD DEL CUADRO CLÍNICO
El cuadro clínico de la AD y de la Sb0 tal es más severo que el de la HSC y el de la Sb+tal. Sin embargo, el espectro clínico de la AD es muy amplio: desde niños que mueren en los primeros años de la vida hasta pacientes que llegan a la quinta o sexta décadas. Todos presentan una anemia hemolítica crónica de intensidad variable, pero el número de CVOD, STA y otras manifestaciones clínicas varían considerablemente.4 Se han realizado estudios para identificar temprano en la vida los factores pronósticos desfavorables, con el objetivo de tomar medidas que disminuyan su incidencia y retrasen o impidan el daño crónico de los órganos.
El Grupo Cooperativo de Estudio de la Drepanocitosis (CSSCD) de los EE.UU. reporta que la dactilitis y la anemia severa en el primer año de la vida y la leucocitosis en el segundo, son factores pronósticos desfavorables que se relacionan con una evolución posterior adversa, que incluye: muerte, AVE, CVOD frecuentes y STA recurrente.12 Este modelo no se ha validado en otra investigación en la que se plantea que solo el conocimiento de la genómica y de la proteómica de la enfermedad podrá predecir su curso.13 Estos estudios no son totalmente comparables. Entre las diferencias más importantes están que el primero se realizó antes de la utilización de la penicilina oral profiláctica e incluyó todas las CVOD; mientras que el segundo solo consideró las CVOD que requirieron hospitalización. Una investigación que incluye una cantidad grande de variables clínicas y de laboratorio llega a la conclusión de que el riesgo de muerte se asocia con la intensidad del proceso hemolítico, con la insuficiencia renal crónica y con la leucocitosis. También el AVE, el STA, la sepsis, las CVOD y el priapismo, son característicos de enfermedad severa. Sin embargo, ningún evento clínico por sí solo, salvo la hipertensión pulmonar, ni ningún dato de laboratorio aislado, predice mortalidad. El genotipo es un factor pronóstico importante, ya que la coexistencia de la AD con a talasemia le confiere al paciente una supervivencia mayor.14 Está claro que hasta el momento actual no se ha podido determinar qué factores pronósticos, temprano en la vida, permitirían identificar con certeza al paciente cuyo curso posterior va a ser grave y realizar tratamientos preventivos y potencialmente curativos, como el trasplante de progenitores hematopoyéticos.
ESTUDIOS REALIZADOS EN EL INSTITUTO DE HEMATOLOGÍA e INMUNOLOGÍA (IHI)
Epidemiología
El comercio de esclavos desde África hacia Cuba produjo un grado variable de mezcla racial que explica la composición genética de la población. En blancos, se encuentra el 5 % de genes negros y en negros, el 13 % de genes blancos. La frecuencia del estado de portador AS en el país es del 0,6 % en blancos; 4,1 % en mestizos; 13 % en negros; y 3,08 % en la población general. Su prevalencia es mayor en las provincias orientales, donde el porcentaje de población negra es mayor. La frecuencia del estado de portador de Hb C es del 0,6 % y la de la b tal heterocigótica del 0,8 %. El 22,7 % de la población no blanca es portadora de a talasemia (aa/-a).15
En el año 1973 se comenzaron a estudiar en el IHI diferentes aspectos clínicos y hematológicos de la drepanocitosis.16-22
Crisis aguda de secuestro esplénico
Es una complicación muy frecuente en el niño menor de 5 años que se define como una disminución brusca de la cifra de Hb de más de 2g/dL por debajo del nivel basal, con aumento doloroso del tamaño del bazo. Si no se trata rápidamente con transfusión de glóbulos rojos, tiene una alta mortalidad. El tratamiento clásico es la esplenectomía total en el niño mayor de 4 años, que tiene el riesgo de sepsis fulminante que conduce a la muerte al 50 % de los pacientes. Desde el año 1986, en el IHI se realiza esplenectomía parcial con excelentes resultados. Hasta el momento actual se han realizado 80 intervenciones. Parte de los resultados de esta investigación se han publicado23 y ninguno de los pacientes operados repitió la crisis y solo uno presentó una meningitis neumocóccica de la que se recuperó sin secuelas.24
INVESTIGACIONES QUE SE DESARROLLAN ACTUALMENTE EN EL IHI
Accidente vascular encefálico (AVE)
Se define como la aparición de manifestaciones neurológicas de más de 24 horas de duración. Es una de las complicaciones más graves de la enfermedad, que puede llevar a la muerte o dejar secuelas importantes. Su frecuencia es de alrededor del 10 % entre los 5-10 años de edad25 y se produce por la estenosis u obstrucción de las arterias cerebrales medias, cerebrales anteriores o carótida interna. Existen factores predisponentes al AVE: ataques isquémicos transitorios, hipertensión arterial, STA previo, anemia severa, leucocitosis. Uno de los factores pronósticos más importante es el aumento de la velocidad del flujo sanguíneo en las arterias cerebrales (VFC) medido por ultrasonido Doppler transcraneal (UDT).26
La manifestación clínica más frecuente es la hemiplejia, pero cualquier manifestación neurológica puede ocurrir. El diagnóstico se realiza por tomografía axial computadorizada en la que las lesiones aparecen aproximadamente 6 horas después de ocurrido el AVE; y por resonancia magnética, en la que las lesiones aparecen 2-4 horas después.
La recurrencia de esta complicación es de alrededor del 70 %, y cada episodio puede conducir a la muerte o agravar las secuelas ya existentes. El tratamiento es la exanguinotransfusion realizada lo más rápidamente posible después del diagnóstico y posteriormente, un régimen sistemático de transfusiones periódicas cuya duración no se ha establecido todavía, aunque es probable que haya que mantenerlo hasta los 18 años de edad para disminuir la recurrencia en aproximadamente el 10 %.25,26
Adams y otros, demostraron que en un número grande de pacientes se puede predecir el AVE. Si la VFC es mayor que 200 cm/seg, la probabilidad de AVE es del 60 %. Una velocidad de 170 cm/seg se considera condicional y debe repetirse el UDT frecuentemente.26
El régimen de transfusiones periódicas tiene muchas complicaciones: aloinmunización, sobrecarga de hierro, infecciones (síndrome de inmunodeficiencia adquirida y hepatitis C, entre otras) y dificultades con el acceso venoso. Por otro lado, no es efectivo en todos los pacientes y la respuesta de la vasculopatía cerebral es variable y no siempre beneficiosa.27
Una alternativa al régimen de transfusiones periódicas es el tratamiento con hidroxiurea, que ha probado ser útil en un grupo de pacientes como prevención primaria y también para evitar las recurrencias.28
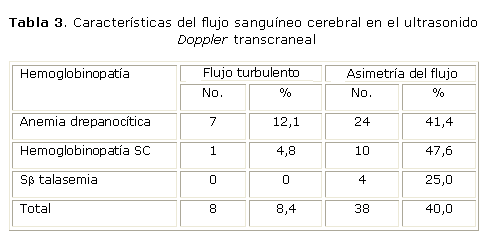
Entre el período de enero 2007- junio 2009, en el IHI se estudió la VFC con un aparato de ultrasonido Doppler color Combimson, Fretz, Austria, en 95 niños en condiciones basales, sin manifestaciones clínicas en el mes anterior y sin transfusiones en los 3 meses previos al estudio; y 20 controles de aproximadamente la misma edad y sexo que los pacientes. En los niños estudiados, la VFC resultó condicional en 7 (6,4 %) y anormal en 5 (4,6 %). En la tabla 1 se muestran la edad y sexo de los pacientes estudiados en los diferentes tipos de drepanocitosis. La VFC en las arterias cerebrales se observa en la tabla 2. La VFC fue mayor en la AD y Sb0 tal que en la HSC y en la Sb+ tal, con diferencias estadísticamente significativas. En la AD, la VFC disminuyó con la edad. No se encontró relación significativa entre los parámetros hematológicos Hb, reticulocitos, leucocitos y Hb fetal (Hb F) y la VFC.29 En los pacientes que sufrieron AVE, el tratamiento fue la exanguinotransfusión y posteriormente un régimen sistemático de transfusiones periódicas para mantener el nivel de Hb S en menos del 30 %; e hidroxiurea 25 mg/k/día durante 1 año. Luego, hidroxiurea en igual dosis durante un tiempo que aún no se ha definido con exactitud. Recientemente se ha planteado que la turbulencia del flujo sanguíneo cerebral en el sitio de la estenosis es un factor que produce más injuria vascular y predispone al AVE.30 En nuestros pacientes se encontró flujo turbulento en el 8,4 % de los casos y asimétrico en el 40 %; a este último dato no se refiere la literatura y no conocemos aún su causa (tabla 3).

La enfermedad cerebrovascular silente es mucho más frecuente que el AVE. Sus diferencias se muestran en la tabla 4. Los factores de riesgo son: pocas CVOD, convulsiones, cifra de leucocitos mayor que 11,8/109/L y el haplotipo Senegal. El diagnóstico se sospecha cuando existen alteraciones neurocognoscitivas y disminución del índice académico y se confirma por medio de la resonancia magnética, que muestra pequeños infartos subcorticales.31
En la tabla 5 se muestran los resultados del estudio neurocognoscitivo por medio de la prueba de Weschler realizado en un grupo de niños con AD atendidos en el IHI, con edades entre 5-18 años. Dicho estudio mostró alteraciones en la escala ejecutiva y en el cociente de inteligencia en relación con un grupo control. En entrevistas realizadas con maestros, el 36 % de los niños tenían dificultades en el aprendizaje del español y el 40 % en el de matemática. Parte de estos resultados se han publicado.32

En el Instituto Nacional de Salud y en el Instituto Nacional de Enfermedades Neurológicas y Accidente Vascular Encefálico de los EE. UU., comenzó un estudio de enfermedad cerebrovascular silente en 1 800 pacientes en 24 centros de ese país, Canadá, Inglaterra y Francia, con una duración de 6,5 años. Los enfermos con una VFC de más de 200 cm/seg se dividirán al azar para recibir transfusiones o solo observación.33 Este estudio es muy importante, porque a los 14 años, el 20 % de los niños con AD padecen de enfermedad cerebrovascular silente.
Alteraciones renales
La insuficiencia renal crónica se presenta en el 20 % de los adultos con AD, pero las evidencias de lesiones renales se encuentran desde muy temprano en la niñez. La patogenia es conocida; la polimerización de la Hb S es mayor en la médula renal, en la que existen hipoxia, acidosis e hipertonía. La microalbuminuria es un signo temprano de daño glomerular que se presenta antes de que disminuya la velocidad del filtrado. Se plantea que las enzimas convertidoras de la angiotensina, así como las transfusiones crónicas y la hidroxiurea, pueden preservar la función renal.34
Entre los años 2005-2008 se estudiaron en el IHI con el reactivo HELFA (Empresa de Productos Biológicos "Carlos J. Finlay", Cuba), 99 pacientes de 5-18 años de edad en condiciones basales. Se realizaron 3 determinaciones de microalbuminuria en el transcurso de 15 días y la prueba se consideró positiva cuando la cifra fue mayor que 0,02 mg/L en 2 de ellas. La positividad de la microalbuminuria en los diferentes tipos de drepanocitosis y hemoglobinopatías, se observa en la tabla 6. Aunque el número de pacientes fue pequeño, es interesante señalar que se encontró correlación entre la positividad de la prueba y el haplotipo Bantú.35

La utilidad de los convertidores de la angiotensina se discute debido a que la microalbuminuria puede ser transitoria e intermitente. Algunos autores indican este tratamiento solo si existe proteinuria (relación albuminuria/creatinina mayor de 300mg/g).34 En todos los niños que presentaron microalbuminuria en nuestro trabajo se utilizó captopril. Los resultados de esta investigación están en proceso de análisis en la actualidad.
Oximetría de pulso
La saturación de oxígeno (SaO2) en la drepanocitosis es más baja que en niños normales debido, en parte, a la disminución de la afinidad por el oxígeno de la Hb S, aunque no se conocen todos los factores que la producen, ya que pacientes con cifras muy bajas de Hb tienen SaO2 normales. La hipoxia es un factor desencadenante de las CVOD, STA y muchas otras manifestaciones de la enfermedad. La oximetría de pulso (OP) es un procedimiento sencillo que refleja adecuadamente la SaO2. Recientemente, se recomienda realizar la SaO2 diurna en condiciones basales en varias oportunidades para comparar estos valores con los obtenidos en algunas complicaciones como el STA. Más importancia que a la OP diurna se le concede al registro nocturno que detecta la apnea obstructiva del sueño y se correlaciona con un aumento de la frecuencia de las CVOD, convulsiones, ataques isquémicos transitorios y AVE.36 Sin embargo, recientemente se ha planteado que la SaO2 diurna se correlaciona con la nocturna y con la apnea obstructiva del sueño, por lo que también es un factor de riesgo para el AVE.37 Para algunos autores, la SaO2 diurna se correlaciona de manera inversa con la VFC en el Doppler transcraneal, factor pronóstico bien conocido en el AVE.38
En el período entre enero 2005-diciembre 2007, se estudió la SaO2 con el aparato OXY9800 (ICID/Combiomed, Cuba) en 108 niños del IHI de 3-18 años de edad, en condiciones basales: 67 con AD y Sb0 tal y 41 con HSC y Sb+ tal; 57 del sexo masculino y 20 controles de aproximadamente la misma edad y sexo. Se consideró hipoxemia una SaO2 igual o menor que el 94 %. Los controles tuvieron una SaO2 promedio del 97,8 %. El promedio en los pacientes con AD y Sb0 tal fue de 93,9 % DE 4,8; y en la HSC y Sb+, de 95,1 % DE 2,4. La desaturación de la Hb fue mayor en el primer grupo con diferencias estadísticamente significativas (tabla 7). En este estudio, la SaO2 en la AD y Sb0 disminuyó con la edad, no así en la HSC y Sb+ tal. Es probable que esto se deba al comienzo de la hipertensión pulmonar en el primer grupo (datos no publicados).

Hipertensión pulmonar
Es una complicación de la drepanocitosis y otras anemias hemolíticas congénitas a la que se le ha dado mucha importancia en los últimos años. Con el aumento de la sobrevida del paciente en las últimas décadas, han aumentado las complicaciones crónicas, entre ellas, la hipertensión pulmonar (HTP). Alrededor del 30 % de los pacientes adultos con AD la presentan. El diagnóstico se realiza por el ecocardiograma que muestra una velocidad de regurgitación en la válvula tricúspide (VRT) de 2,5 m/seg o más, un umbral que se corresponde con una presión sistólica en la arteria pulmonar de 30 mm Hg.39 Aunque estas cifras expresan una HTP ligera o moderada, en la AD es grave, ya que el 40 % de los enfermos mueren en los 40 meses posteriores al diagnóstico.40
En 600 niños en los que se investigó HTP, se encontró una incidencia semejante a la del adulto y se acompañó de proteinuria y un estado protrombótico y proinflamatorio, posiblemente favorecido por la asplenia funcional.40
Las manifestaciones clínicas más frecuentes de la HTP, aunque tardías, son la disnea de esfuerzo y los dedos en palillo de tambor.40
La deshidrogenasa láctica (LDH) es un marcador biológico de la resistencia al óxido nítrico (ON) asociada con la hemólisis y se debe realizar para detectar precozmente esta complicación.41
Se han propuesto varios tratamientos para revertir la HTP o retrasar su progresión: régimen sistemático de transfusiones periódicas, hidroxiurea utilizada desde temprano en la vida, prostanoides IV, L-Arginina y Bosentan (inhibidor de la endotelina-1). Hasta el momento, el más útil parece ser el sildenafil, inhibidor de la fosfodiestearasa tipo 5.41
En el IHI se está realizando ecocardiograma y LDH a todos los niños entre 3-18 años. Hasta el presente se han estudiado 25 con AD, entre quienes se encontraron 6 (25 %) con VRT mayor de 2,5 m/seg. El número de pacientes estudiados es aún muy pequeño, pero el porcentaje de HTP es semejante al reportado en la literatura.40
Tratamiento con hidroxiurea (HU)
En los últimos años se han propuesto múltiples tratamientos destinados a aumentar el porcentaje de Hb F, disminuir la polimerización de la Hb S, mejorar la hidratación del hematíe, disminuir la adhesión de los hematíes y leucocitos al endotelio vascular, mejorar la reología de la sangre, disminuir la vasoconstricción y otros. El único que ha probado su eficacia en ensayos clínicos controlados en niños y adultos es la hidroxiurea, un citotóxico inhibidor de la ribonucleótido sintetasa que aumenta la Hb F y el número de células F, disminuye la hemólisis, la producción de leucocitos y de reticulocitos de estrés, inhibe la depleción de cationes y por lo tanto, mejora la hidratación del hematíe, aumenta el ON, disminuye la producción de moléculas de adhesión y aumenta la deformidad del glóbulo rojo, con lo que mejora la reología de la sangre.42
En el IHI se trataron con HU en el período comprendido entre junio 2003 y junio 2007: 51 niños con drepanocitosis; 39 con AD; 7 con HSC; y 5 con Sb tal, todos con una mediana de edad de 10 años (3-17), una mediana de seguimiento de 25 meses (4-57); 43 % del sexo masculino. Los criterios de inclusión fueron 3 o más CVOD al año en los 3 años previos; uno o más episodios de STA en los 2 años anteriores; y un AVE en el último año. El seguimiento se realizó mensualmente con hemograma y reticulocitos en cada consulta y alaninoaminotransferasa, creatinina y Hb F cada 3 meses. La dosis utilizada fue menor que la que recomienda la literatura: 15 mg/kg/día. Las manifestaciones clínicas disminuyeron significativamente (tabla 8). Se logró también un aumento significativo de la Hb total y de la Hb F y una disminución de los reticulocitos (tabla 9); parte de esta investigación se ha publicado.43 Iguales resultados se obtuvieron en un estudio realizado en colaboración con algunos países de Centroamérica y el Caribe.44


ATENCIÓN INTEGRAL AL PACIENTE CON DREPANOCITOSIS
En 1974, se realizó en Cuba un taller en el que se abordaron por distintas instituciones (IHI, Departamento de Genética Médica del Instituto de Ciencias Básicas y Preclínicas "Victoria de Girón", el Grupo de Genética Médica de la provincia de Villa Clara y el Ministerio de Salud Pública), las proyecciones para establecer y desarrollar un programa preventivo nacional para la drepanocitosis, de alcance poblacional.45 El programa se basó en la identificación de las parejas portadoras de Hb S o Hb C, la opción de diagnóstico prenatal, el asesoramiento genético y la posibilidad de interrupción del embarazo si la pareja así lo decidía, o la identificación del niño enfermo antes de su nacimiento para su inclusión en el programa de atención integral. El diagnóstico se realizó por medio del estudio del ADN fetal que permite distinguir entre el feto sano, el portador o el enfermo. Entre los años 1982-1986 y con la introducción de un sistema de electroforesis desarrollado y patentado por investigadores cubanos,46 se comenzó gradualmente a poner en práctica el programa en diferentes provincias, y en el año 1988 se extendió a todo el país.47 En el 82 % de las parejas de las gestantes portadoras de Hb S o Hb C se realizó la electroforesis de Hb. Los registros estadísticos y el control del programa se llevaron a cabo en los centros provinciales de genética médica y el estudio del ADN del feto en el Centro Nacional de Genética Médica. Los resultados del plan de diagnóstico prenatal en el período diciembre 1982-diciembre 2008 fueron:
- Gestantes estudiadas: 3 475 931.
- Gestantes portadoras: 124 664 (3,58 %).
- Parejas de riesgo: 5 640.
- Diagnóstico prenatal molecular: 4 033.
- Fetos enfermos: 800.
- Embarazos interrumpidos: 637.
Se estima que en la actualidad la enfermedad ha disminuido de manera considerable en el país. De alrededor de 100 nacimientos de niños enfermos anuales, nacen actualmente un promedio de 10, en su mayoría descendientes de parejas que aunque conocían el diagnóstico en la etapa prenatal, decidieron continuar el embarazo (Dra. Beatriz Marcheco, comunicación personal).
En Cuba existe un plan de atención integral que incluye el diagnóstico prenatal y el seguimiento en consultas especializadas desde poco después del nacimiento.
En 1990 se creó en el IHI una consulta de hemoglobinopatías en la que se atiende a los pacientes cada 3 meses desde los primeros meses de la vida. En esta consulta se realiza un interrogatorio sobre las manifestaciones clínicas ocurridas en el período entre las visitas y un hemograma completo y recuento de reticulocitos. Se administra penicilina oral profiláctica desde los 3 meses hasta los 5 años de edad. Se recomienda al paciente la ingestión abundante de líquidos, 1mg de ácido fólico al día toda la vida, medidas higiénico-dietéticas, evitar esfuerzos físicos excesivos (sobre todo la intervención en competencias deportivas) y se estudia su crecimiento y desarrollo. En cada consulta se instruye a la madre y al padre para que conozcan el color de la piel del niño y aprendan a palpar el bazo. Existen normas de tratamiento y un folleto explicativo para pacientes y familiares que se han distribuido a todo el país. Se administra consejo genético y orientación vocacional y cuando es necesario, se establecen relaciones con la escuela o el centro de trabajo. Con todas estas medidas, se ha logrado prolongar la vida y mejorar su calidad. En el período 2004-2008 han fallecido 16 niños en todo el país (datos del Ministerio de Salud Pública, junio 2009); de ellos, 4 en el IHI: 2 por STA, 1 por insuficiencia renal crónica y 1 por sepsis generalizada.
La supervivencia en 397 adultos fue de 53 años en la AD y 58 en la HSC.21
Un plan de atención integral en los países subdesarrollados es muy importante porque permite prolongar la supervivencia y mejorar la calidad de vida del paciente. Además, tiene la ventaja de que disminuye el gasto ocasionado por su atención, ya que el tratamiento de las complicaciones es mucho más costoso que el de las medidas necesarias para su prevención.
De acuerdo con todos los datos antes expuestos, algunos de los resultados más importantes que se deben destacar son:
1. Conocimiento de la prevalencia del estado de portador en los diferentes grupos de población y en diferentes lugares del país.
2. Desarrollo del programa nacional de diagnóstico prenatal de la AD y de la HSC, con el que se logró reducir de manera significativa el nacimiento de niños enfermos.
3. Creación de una consulta especializada y un plan de atención integral, con el que se ha logrado disminuir la mortalidad en niños y adultos y mejorar su calidad de vida.
4. Tratamiento con hidroxiurea en la AD que aumentó la Hb y la Hb F y disminuyó significativamente la frecuencia de CVOD, STA, transfusiones y hospitalizaciones.
5. Detección de los pacientes con VFC condicional (mayor de 170 cm) o anormal (mayor de 200 cm) y la prevención del AVE con la administración de hidroxiurea.
6. Tratamiento con captopril en los niños con microalbuminuria para disminuir la incidencia de insuficiencia renal crónica.
7. Utilidad de la esplenectomía parcial en la CSE.
8. Estudio neurocognoscitivo que mostró deficiencias en la escala ejecutiva y en el cociente de inteligencia.
9. Determinación de la SaO2 diurna que mostró una disminución más marcada en la AD y Sb0 tal que en la HSC y en la Sb+tal.
10. Estudio de la HTP en niños con AD, que mostró un aumento de la VRT en el 25 % de los pacientes.
REFERENCIAS BIBLIOGRÁFICAS
1. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: An increasing global health problem. Bull WHO. 2001;79:1-15.
2. Roberts I, De Montalembert M. Sickle cell disease as a paradigm of immigration hematology: New challenges for hematologists in Europe. Haematologica 2007;92:865-79.
3. Sergeant GR, Sergeant BC. Sickle cell disease. 3rd ed. New York: Oxford Univ Press; 2001. p. 16-25.
4. Colombo B, Svarch E, Martínez G. Genética y clínica de las hemoglobinas humanas. La Habana: Ed Pueblo y Educación; 1993. p. 146-83.
5. Svarch E. Fisiopatología de la drepanocitosis. Rev Cubana Hematol Inmunol Hemoter [serie en internet]. 2009;25(1). [citado 29 octubre 2009]; Disponible en: http://bvs.sld.cu/revistas/hih/vol25_1_09/hih03109.htm
6. Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol.
2009;84:618-25.
7. Chiang EY, Frenett PS. Sickle cell vaso-occlusion. Hematol Oncol Clin North Am 2005;19:771-84.
8. Nathan DG, Orkin SH, Ginsburg D, Look AT. Hematology of infancy and childhood. 6th ed. Philadelphia: Sounders; 2003. p. 790-841.
9. Vishinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D, et al. Causes and outcomes of the chest syndrome in sickle cell disease. N Eng J Med. 2000;342:1855-65.
10. Chaar V, Tarer V, Etienne-Julan M, Dlara JP, Elion J, Romana M. ET-1 and ec NOS gene polymorphism and suceptibility to acute chest syndrome and painful vaso-occlusive crisis in children with sickle cell anemia. Haematologica 2006;91:1227-8.
11. Kato J, Gladwin MT, Steinberg MH. Deconstructin sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37-47.
12. Miller ST, Sleeper LA, Pegelow CH, Enos MC, Wang WC, Weiner SJ, et al. Prediction of adverse outcomes in children with sickle cell disease. N Eng J Med. 2000;342:83-9.
13. Quinn CT, Lee NJ, Rogers ZR, Buchanan GR. Prediction of adverse outcomes in children with sickle cell anemia: a study of the Dallas newborn cohort. Blood. 2008;111:544-8.
14. Sebastiani P, Nolan VG, Baldwin CT, Abad-Grau MM, Wang L, Adewoye AH, et al. A network model to predict risk of death in sickle cell disease. Blood. 2007;110;2727:35.
15. Colombo B, Martínez G. Hemoglobin variants in Cuba. Hemoglobin. 1985;9:415-22.
16. Menéndez A, Svarch E. La crisis aplástica en las anemias hemolíticas congénitas. Rev Cubana Hematol Inmunol Hemoter. 1987;3:125-30.
17. Sagarra M, Svarch E, Hernández P. Alteraciones óseas en las hemoglobinopatías. Rev Cubana Med. 1984;601:5-8.
18. Svarch E, González A, García T. Anemia drepanocítica. Estudio de 110 pacientes. Rev Cubana Ped. 1986;56:3-9.
19. Svarch E. Plasma Exchange for acute cholestasis in homozygous sickle cell disease. Haematologica. 1986;19:49-52.
20. García T, Nordet I, Machin S, González A, Muñiz A, Martínez G, et al. La drepanocitosis en los primeros 5 años de la vida. Rev Cubana Hematol Inmunol Hemoter. 1999;15:96-104.
21. Machín S, Guerra T, Svarch E, Espinosa E, Mesa J, Dorticós E, et al. Morbiletalidad en pacientes adultos con drepanocitosis. Rev Cubana Hematol Inmunol Hemoter 2004;20. [citado 29 octubre 2009]; Disponible en:
http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892004000200004&lng=es&nrm=iso&tlng=es
22. Espinosa E, Svarch E, Martínez G, Hernández P. La anemia drepanocítica en Cuba. Experiencia de 30 años. Rev Cubana Hematol Inmunol Hemoter. 1997;12:1-8.
23. Svarch E, Nordet I, Valdés J, González A, Machin S, De la Torre E. Partial splenectomy in children with sickle cell disease. Haematologica. 2003;88:222-3.
24. Svarch E, Nordet I, González A. Overhelming septicaemia in a patient with sickle cell/b0thalassaemia. Brit J Haematol. 1999;104:930.
25. Ohene-Frempong K. Stroke in sickle cell disease: Demographic,clinical and therapeutic considerations. Sem Hematol. 1991;28:213-9.
26. Adams RJ, Mc Kie VC, Hsu L, Files B, Vishinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcraneal Doppler ultrasonography. N Eng J Med. 1998;339:5-11.
27. Bader-Meunier B, Verlhac S, Elmaleh-Berges M, Ithier G, Sellami F, Faid S, et al. Effect of transfusion on cerebral vasculopathy in children with sickle-cell anemia. Haematologica. 2009;94:123-6.
28. Gulbis B, Haberman D, Dufour D, Christophe C, Vermylen C, Kagambega F, et al. Hydroxyurea for sickle cell disease for prevention of cerebrovascular events: The Belgian experience. Blood. 2005;105:2685: 90.
29. Menéndez A, Scherer C, Mesa F, Arencibia A, Nordet I, Pozo A, et al. Ultrasonografía doppler transcraneal en niños con drepanocitosis. Rev Cubana Hematol Inmunol Hemoter [serie en internet]. 2009 Abr [citado 29 octubre 2009]; 25 (Sup): Disponible en: http://bvs.sld.cu/revistas/hih/vol25_04_09/hih05409.htm
30. Quinn CT, Variste J, Dowling MM. Haemoglobin oxygen saturation is a determinant of cerebral artery blood flow velocity in children with sickle cell aneamia. B Med J. 2009;145:500-5.
31. Kirkham FJ, Lerner NB, Noetzel M, De Baum MR, Datta AK, Rees DC, et al. Trials in sickle cell disease. Ped Neurol. 2006;34:450-8.
32. Martínez R, Svarch E, Menéndez A. Limitación cognitiva en niños con anemia drepanocítica sin historia de afectación neurológica. Rev Cubana Hematol Inmunol Hemoter [serie en internet]. 2009;25: [citado 29 octubre 2009]; Disponible en:
http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892009000100009&lng=es&nrm=iso&tlng=es
33. Nabel EG, Shurin SB. A recommitment to sickle cell disease research. Blood. 2008;111:4852-3.
34. Álvarez O, López-Mitnik G, Zilleruelo G. Short term follow-up of patients with sickle cell disease and albuminuria. Pediatr Blood Cancer. 2008;50:1236-9.
35. Ramos AI. Prevalencia de microalbuminuria en niños con drepanocitosis y respuesta al captopril. [Tesis de especialidad]. Ciudad de La Habana, Cuba. ISCMH; 2010.
36. Kirkham FJ, Hewes DK, Pregler M, Lane R, Evans JP. Nocturnal hypoxaemia and central-nervous-system events in sickle cell disease. Lancet. 2001;357:1565-9.
37. Spivey JF, Uong EC, Strunk R, Boslaugh SE, De Baum MR. Low daytime pulse oximetry reading is associated with nocturnal desaturation and obstructive sleep apnea in children with sickle cell anemia. Pediatr Blood Cancer. 2008;50:359-62.
38. Quinn CT, Sargent JW. Daytime steady-state haemoglobin desaturation is a risk factor for overt stroke in children with sickle cell anaemia. Br J Haematol. 2008;140:336-9.
39. Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk for death in patients with sickle cell disease. N Eng J Med. 2004;350:886-95.
40. Kato GJ, Onyekwere OC, Gladwin MT. Pulmonary hypertension in sickle cell disease. Relevance in children. Pediatr Blood Cancer. 2007;24:159-70.
41. Kato GJ, Mc Gowan V, Machado RF, Little JA, Taylor VI, Morris CR, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006;107:2279-85.
42. Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Eng J Med 2008;358:1362-9.
43. Machín S, Svarch E, Agramonte O, Núñez A, Menéndez A, Hernández C, et al. Tratamiento con dosis moderadas de hidroxiurea en la drepanocitosis. Rev Cubana Hematol Inmunol Hemoter [serie en internet]. 2008;24. [citado 6 noviembre 2009]; Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892008000200008&lng=es&nrm=iso&tlng=es
44. Svarch E, Machín S, Nieves RM, Mancía de Reyes AG, Navarrete M, Rodríguez H. Hydroxyurea treatment in children with sickle cell disease in Central América and the Caribbean countries. Pediatr Blood Cancer. 2006;47:111-2.
45.De la Torre E, Colombo B, Hernández P, Heredero L, Svarch E. Taller de sicklemia. Rev Cubana Pediatr. 1974;46:249-61.
46.Heredero L, Granda H, Suárez JA, Atland K. An economic high-speed electrophoretic screening system for hemoglobin S and other proteins. Humangenetik. 1974;21:167-77.
47.Granda H, Gispert S, Martínez G, Gómez M, Ferreira R, Collazo T, et al. Results from a reference laboratory for prenatal diagnosis of sickle cell disorder in Cuba. Prenatal Diagnosis. 1994;14:659-62.
Recibido: 12 de agosto del 2010.
Aprobado: 30 de agosto del 2010.
Prof. DraC. Eva Svarch. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800. Ciudad de La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. correo electrónico: ihidir@hemato.sld.cu; ihi@infomed.sld.cu
Website: http://www.sld.cu/sitios/ihi/index.phpi


{kind=link}
{kind=link}