










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.30 no.2 Ciudad de la Habana abr.-jun. 2014
ARTÍCULO ORIGINAL
Morbilidad y mortalidad de la hemoglobinopatía sc en el Instituto de Hematología e Inmunología. Experiencia de 36 años
Morbility and mortality of hemoglobinopathy sc at the Institute of Hematology and Immunology. Experience of 36 years
Dr. Sergio Machín García, Dra.MirelysCutiño Martínez, Prof. DraC. Eva Svarch, Dr. Alberto Arencibia Núñez, Dra. Andrea Menéndez Veitía, Dr.Carlos Hernández Padrón, Dra. Adys Gutiérrez Díaz, Dra. Rosa M. Lam Díaz
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
Introducción: la hemoglobinopatía SC es la segunda variante de la drepanocitosis más frecuente en Cuba; sin embargo, existen pocos trabajos dirigidos a su estudio.
Objetivo: realizar una caracterización de la historia natural de lahemoglobinopatía HSC,
Métodos: se realizó un estudio observacional, descriptivo, retrospectivo y longitudinal en 148 pacientes con hemoglobinopatía HSC seguidos al menos 2 años en el Instituto de Hematología e Inmunología (1973-2009). Se definieron los eventos hematológicos según las normas cubanas de la drepanocitosis y los exámenes complementarios se realizaron en condiciones basales del paciente. Se utilizó la prueba de Chi Cuadrado para determinar asociación entre variables. Los parámetros de laboratorio se compararon mediante la prueba t Student. Para la estimación de la sobrevida global (SG) se empleó el método de Kaplan Meier.
Resultados: predominó el sexo femenino (56,1 %). La crisis vasoclusiva dolorosa (91,2 %) y el síndrome torácico agudo (35,1 %) fueron las manifestaciones clínicas más frecuentes. El 10,8 % presentó afecciones oftalmológicas (hemovítreo, retinopatía, desprendimiento de retina y catarata). La esplenomegalia predominó en los pacientes menores de 40 años y la hepatomegalia se encontró en todas las edades. Hubo 36 mujeres con embarazos sin mortalidad materna ni perinatal y 26 abortos (65,4 % fueron espontáneos). La anemia fue ligera pero más acentuada en el sexo femenino. Las funciones hepática y renal, mostraron deterioro con la edad. La supervivencia global a los 50 años fue del 79 %. La causa de muerte más frecuente fue la insuficiencia renal crónica.
Conclusiones: el aumento de la calidad y expectativa de vida de la HSC en Cuba es el resultado de la atención médica multidisciplinaria y el fácil acceso a los servicios de urgencia.
Palabras clave: hemoglobinopatía SC, morbilidad, supervivencia.
ABSTRACT
Introduction: hemoglobinophatySC (HSC) is the second most common variant of sickle cell disease in Cuba and the world; nevertheless, there are few studies aimed in this field.
Objective: to make the characterization of the natural history of HSC.
Methods: an observational, descriptive, retrospective and longitudinal study was performed in 148 patients with HSC followed for at least two years at the Institute of Hematology and Immunology in the period 1973-2009. Hematological events according to Cuban procedures in sickle cell disease were determined and complementary studies were performed.
Results: there was a predominance of females (56.1 %). Vasocclusive painful crises (91.2 %) and acute chest syndrome (35.1 %) were the most frequent clinical events. Ophthalmology affections were present in 10,8 % (hemovitreous, retinopathy, retinal detachment and cataract). Splenomegaly was predominant in patients under 40 years and hepatomegaly was found in all ages. There were 36 women with pregnancies without maternal or perinatal mortality. From 26 abortions (65.4 % were spontaneous). Anemia was mild but more pronounced in females. Liver and kidney functions showed deterioration with age. Overall survival at 50 years was 79 %. The main cause of death was chronic renal failure.
Conclusions: increasing the quality of life and life expectancy of HSC in Cuba is the result of multidisciplinary comprehensive care and easy access to emergency services.
Keywords: hemoglobinophatySC, morbility, survival.
INTRODUCCIÓN
La drepanocitosis es la anemia hemolítica crónica hereditaria más frecuente en el mundo. Es multisistémica y cursa con episodios agudos y daño progresivo de órganos.1 Se caracteriza por la presencia de hematíes en forma de hoz (falciforme) en la sangre periférica, que son en gran medida los responsables de las manifestaciones clínicas de la enfermedad.
La drepanocitosis es característica en los afrodescendientesy en aquellas poblaciones en las cuales existe una elevada mezcla racial. Es la hemoglobinopatía más frecuente en los Estados Unidos, en muchos países de América Central y el Caribe, y algunos de América del Sur. También se considera ya un problema de salud en Europa, debido a un aumento considerable de su frecuencia en los últimos 15 años por la emigración desde países en los que la enfermedad es prevalente.2
La frecuencia del estado de portador AS es del 7 % en el mundo y cada año nacen más de 330 000 niños afectados.3
El comercio de esclavos desde África hacia Cuba produjo un grado variable de mezcla racial que explica la composición genética de la población. En blancos, se encuentra el 5 % de genes negros y en negros, el 13 % de genes blancos. La frecuencia del estado de portador AS en Cuba es del 0,6 % en blancos; 4,1 % en mestizos; 13 % en negros; y 3,08 % en la población general. Su prevalencia es mayor en las provincias orientales, donde el porcentaje de población negra es superior. La frecuencia del estado de portador de Hb C es del 0,6 %.4
La hemoglobinopatía SC (HSC) constituye un síndrome doble heterocigotocon patrón de herencia autosómico recesivo, resultado de la herencia del gen bs de un padre y del gen bc del otro progenitor (la HbC se caracteriza por la sustitución del ácido glutámico por lisina en la posición 6 de la cadena b de la Hb).
El cuadro clínico es muy variable entre un individuo y otro e, incluso, en un mismo individuo en diferentes períodos de su vida. Siempre hay hemólisis, pero a diferencia de la anemia drepanocítica (AD), la anemia es a menudo menos severa, de modo que el paciente no suele acudir a los servicios de salud por un problema hematológico. De hecho, en el primer año de vida los niños no presentan síntomas, lo que explica que el diagnóstico resulte más tardío. Aunque la HSC tiene un curso más benigno que la AD, no está exenta de complicaciones, especialmente durante el embarazo y el puerperio. La necrosis aséptica ósea y las afecciones oculares (retinopatía proliferativa, hemorragia del vítreo y desprendimiento de la retina) son más frecuentes que en la AD.5,6
En el año 1960, el pronóstico de la drepanocitosis era tan desfavorable que llegó a ser considerada una enfermedad de la infancia.7 Sin embargo, en las últimas décadas se ha observado un incremento considerable en la expectativa de vida. Estudios realizados en el Instituto de Hematología e Inmunología (IHI) revelan una sobrevida de 58 años en la HSC.8
El desarrollo del Programa Nacional de Prevención de la Drepanocitosis con la aplicación consecuente del diagnóstico prenatal, permite la interrupción del embarazosi la pareja así lo decide, y puede logrardespués de muchos años, una disminución de la incidencia de la enfermedad. Si el embarazo continúa, el seguimiento desde los primeros meses de la vida, que también se logra con el diagnóstico neonatal, conduce a una mayor supervivencia y a una mejor calidad de vida a través de medidas profilácticas y la educación de los pacientes y familiares.
Tanto en Cuba como a nivel internacional, la AD ha sido profundamente estudiada, especialmente su variabilidad clínica, el comportamiento de los parámetros hematológicos y la respuesta a las nuevas alternativas terapéuticas, no así la HSC.
Esta investigación se propuso realizar una caracterización de la HSC en el IHI para mejorar el conocimiento de la historia natural de la enfermedad, perfeccionar el diagnóstico y tratamiento de sus complicaciones e incrementar la calidad y expectativa de vida de los pacientes.
MÉTODOS
Se realizó un estudio observacional, descriptivo, retrospectivo y longitudinal de los pacientes con HSC en el IHI, en el período comprendido entre enero de 1973 y diciembre del 2009.
El universo (N) incluyó 612 pacientes con drepanocitosis atendidos en la consulta externa de hemoglobinopatías del IHI. La muestra (n) se seleccionó según criterios de inclusión y exclusión, para un total de 148 pacientes. Se incluyeron aquellos con diagnóstico de HSC confirmado, seguidos en la consulta externa de la institución al menos durante 2 años y se excluyeron los enfermos con datos incompletos en las historias clínicas.
La información de las variables objeto de estudio se obtuvo de las historias clínicas mediante una planilla confeccionada al efecto. En los casos de fallecimiento se revisó el certificado de defunción y el resultado del estudio anatomopatológico, siempre que estuvieron disponibles.
El proyecto de esta investigación fue aprobado por el Consejo científico y el Comité de ética de la institución. Dado que el estudio se realizó a partir del análisis de la historia clínica, no fue necesario el consentimiento de los pacientes para participar en la investigación. Se respetaron los criterios de confidencialidady los datos obtenidos se emplearon para beneficio científico de interés institucional y de los pacientes.
Se definieron los eventos hematológicos según las normas cubanas de la drepanocitosis.9
Los exámenes complementarios se realizaron en condiciones basales del paciente, sin crisis ni transfusiones en los 3 meses previos. Para el estudio de la hemoglobina fetal (HbF) se empleó el método de la desnaturalización alcalina (Betke) y para los reticulocitos, la tinción con azul brillante de crecilo.
Como medida de resumen para las variables cualitativas se utilizaron las frecuencias absolutas y los porcentajes; y para las cuantitativas, la media y la desviación estándar. Se utilizó la prueba de Chi Cuadrado para determinar asociación entre variables. Los parámetros de laboratorio se compararon mediante la t Student. Para determinar si existían diferencias estadísticamente significativas en las crisis vasoclusivas dolorosa (CVOD) y el síndrome torácico agudo (STA), en los cuatro grupos de edades se utilizó el ANOVA; y para las comparaciones a posteriori se empleó la prueba de Scheffé.
Para la estimación de la sobrevida global (SG) se empleó el método de Kaplan Meier. La comparación de la SG entre los sexos se realizó utilizando la prueba de Breslow, teniendo en cuenta la existencia de entrecruzamiento de las curvas. En todos los casos el nivel de significación estadístico fue 0.05.
RESULTADOS
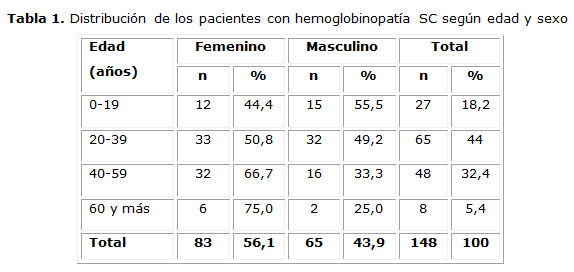
En la tabla 1 se observa la distribución de la muestra por sexo y grupos de edades. Predominó el sexo femenino y los pacientes entre 20-39 años.
De los 113 pacientes en los cuales pudo conocerse la edad del diagnóstico, 50 (33,8 %) fueron diagnosticados después de los 10 años de edad.
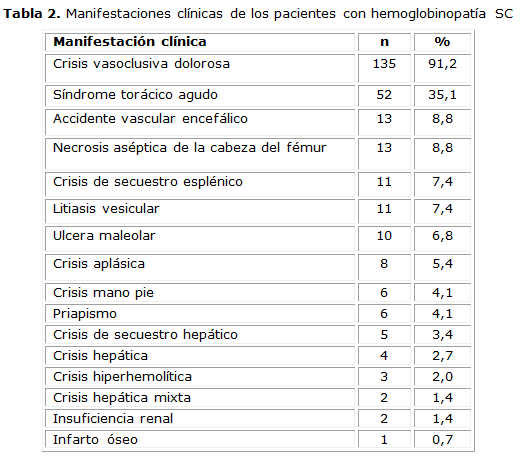
Las manifestaciones clínicas más frecuentes fueron: la CVOD (91,2 %), el STA (35,1 %), el accidente vascular encefálico (AVE) (8,8 %) y la necrosis aséptica del fémur (NACF) (8,8 %). El resto de las crisis se presentaron de manera aislada (tabla 2).
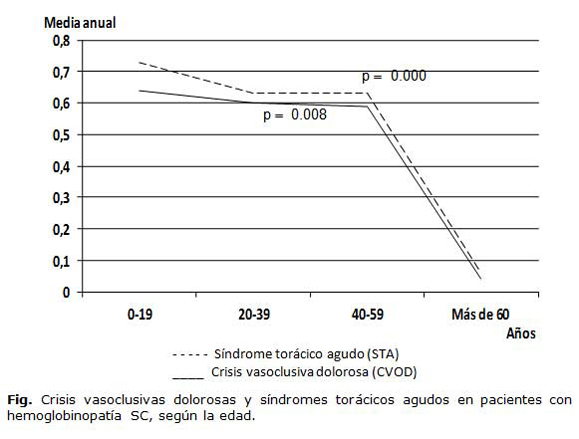
En la figura se presenta la media de CVOD y STA por grupos de edades. Se observó una disminución estadísticamente significativa de ambos eventos con el incremento de la edad (p=0.008 y 0.000, respectivamente).
Se observó una ligera disminución no estadísticamente significativa del promedio de ingresos hospitalarios en los mayores de 60 años. No hubo cambios significativos en el número de consultas entre los diferentes grupos etarios.
El 10,8 presentó afecciones oftalmológicas que incluyeron hemovítreo, retinopatía, desprendimiento de retina y cataratas, sobretodo en mayores de 40 años; 5,4 % refirió padecer de gastritis; 5,4 % hipertensión arterial y 4,1 % asma bronquial.
La esplenomegalia predominó de manera estadísticamente significativa (p=,0.015) en los pacientes con edades entre 0-19 (59,3 %) y 20-39 (60 %). La hepatomegalia se encontró en todos los grupos etarios, sin diferencias estadísticamente significativas.
De 71 mujeres mayores de 19 años, 36 (50,7 %) se embarazaron al menos una vez, para un total de 65 embarazos: 39 partos (60 %) y 26 interrupciones (40 %). De estos últimos, 17 (65,4 %) fueron espontáneos. No hubo muerte materna ni perinatal.
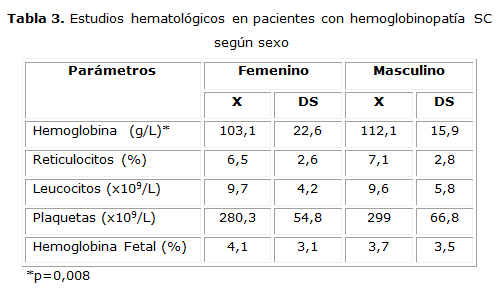
En la tabla 3 se describen los resultados de los estudios hematológicos para ambos sexos. Solo se encontró significación estadística en la hemoglobina, que fue superior en el sexo masculino (p = 0.008). No hubo diferencias en el resto de los parámetros.
De los 108 pacientes que se realizaron estudios bioquímicos, solo se observaron alteraciones en los mayores de 40 años: 2 pacientes presentaron cifras elevadas de creatinina, ambos asociados con insuficiencia renal crónica (IRC), y 4 tuvieron valores aumentados de alaninoaminotransferasa (ALT).
La SG a los 50 años fue del 62,6 %. No hubo diferencia estadística significativa en la probabilidad de supervivencia entre los sexos, aunque fue mayor en las mujeres que mostraron a los 50 años una probabilidad de supervivencia del 60,8 % y 61,7 % los hombres.
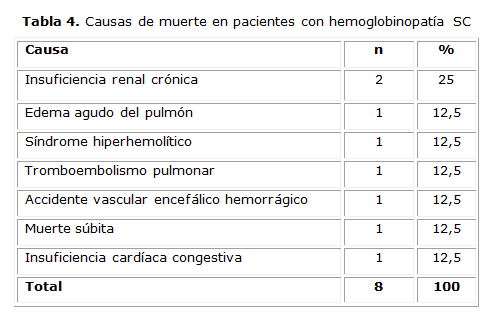
Fallecieron 8 pacientes (5,4 %) con un promedio de edad de 32 años. La primera causa de muerte fue la IRC (tabla 4).
DISCUSIÓN
Este estudio mostró un predominio de pacientes con edades comprendidas entre 20-39 y 40-59 años, este último considerado como el grupo de edad avanzada en diferentes artículos.10-12 Resulta además interesante que el 5,4 % tenía 60 años y más, lo que nos permite afirmar que la HSC tiene una larga sobrevida. Otros trabajos reportan grupos de pacientes con más de 70 años de edad. Este hecho ha motivado, incluso, nuevos estudios en pacientes con edad avanzada, como el realizado en Trinidad y Tobago en el 2006 que incluyó a 40 pacientes mayores de 40 años.13
También se encontró un predominio del sexo femenino, lo cual puede estar relacionado con un mayor interés de la mujer en asistir a consulta de seguimiento y la remisión a la institución de todas las embarazadas con HSC desde los diferentes centros de salud de la ciudad para su seguimiento multidisciplinario.
El hecho de que 50 (33,8 %) de los 113 pacientes en los cuales pudo conocerse la edad del diagnóstico fueran diagnosticados después de los 10 años de edad, habla a favor de un cuadro clínico ligero y coincide con el criterio de otros autores. Según se comenta en la literatura, el diagnóstico de la HSC es más tardío que en la AD. En el primer año de la vida los niños no suelen presentar síntomas estos aparecen en el 50 % entre 1-5 años de edad; el 22 % permanecen asintomáticos hasta los 10 años y en muchos casos el diagnóstico se lleva a cabo por la presencia de una esplenomegalia encontrada en un examen de rutina, o en el curso del embarazo. Al contrario de lo que sucede en la AD, los síntomas más importantes de la HSC por lo general no aparecen hasta la adolescencia.14
Una de las investigaciones más relevantes sobre la clínica de los pacientes con HSC que se ha llevado a cabo hasta hoy, fue el estudio realizado en la universidad del sur de California en el año 2002, que incluyó a 284 pacientes y se extendió por un período de 4 décadas.15
En el presente estudio, que abarcó 36 años, se encontró que el promedio de CVOD fue de 91,2 %, cifra superior a la descrita en el estudio antes citado, donde plantean que el 55 % presentó al menos un episodio doloroso.15Sin embargo, los STA se comportaron de manera similar en ambos estudios (35,1 vs. 32 %). Estos pacientes cursan con una función pulmonar normal durante la infancia que luego tiende a ir declinando paulatinamente.16
El porcentaje de pacientes con hospitalizacionesfue similar al que se describe en la literatura (71 %).15
Las frecuencias de CVOD, STA e ingresos son considerados indicadores de la severidad clínica en la drepanocitosis.17En el caso particular de las CVOD, si su frecuencia es superior a 3 por año, constituye un factor de riesgo de muerte precoz.18 La media de CVOD y STA fue de menos de 1 por año y disminuyeron con la edad. Estos elementos permiten plantear que los pacientes con HSC estudiados se caracterizan por tener un cuadro clínico ligero, especialmente los mayores de 60 años, que coincide con lo que se describe en la literatura.13,15
Una de las complicaciones más frecuentes en la HSC son las alteraciones oftalmológicas. En el 10,8 % de los enfermos se describen hemovítreo, catarata, desprendimiento de retina y retinopatía. Un comportamiento similar se refleja en estudios de grandes series que reportan hasta el 23 % de retinopatías en pacientes con HSC. La retinopatía proliferativa es más frecuente que en la AD.15 El evento inicial en la patogénesis de la retinopatía proliferativa es la oclusión de arteriolas de la retina periférica que requiere un seguimiento estricto por la posibilidad de pérdida mantenida de la visión. Se estima que la prevalencia es mayor en la segunda mitad de la tercera década de la vida, en el sexo masculino y en individuos con bajos niveles de HbF.19
La afectación de los pacientes por asma bronquial (4,1 %) fue inferior a los reportes en la AD. Algunos estudios hablan de una prevalencia del 30-70 % en la AD.20,21 En un estudio de 1963 pacientes con drepanocitosis en Estados Unidos en el 2007,Boydet al.describieron un riesgo de muerte incrementado en aquellos que padecían de asma bronquial. No se encontraron reportes que hablaran sobre la frecuencia de asociación asma bronquial y HSC.
La relación del asma con un aumento de la frecuencia de los STA y el riesgo de muerte en los pacientes con drepanocitosises un aspecto que aún se encuentra en investigación.22,23
En este estudio se encontró un predominio de la esplenomegalia en el adolescente y eladulto joven. Esto quizás pueda explicarse porque los pacientes con HSC tienen menos fenómenos de oclusión vascular. Ello justifica, además, que los pacientes con HSC tengan su primer secuestro esplénico en edades posteriores a los 5 años e incluso en la adultez, debido a un aumento de la susceptibilidad por la persistencia de la esplenomegalia. 24,25A menudo el hallazgo de esplenomegalia moderada en un examen de rutina constituye el punto de partida para el diagnóstico de una HSC.
La hepatomegalia se constató en todos los grupos de edades, tal como aparece descrito en la literatura.1
La literatura señala que las gestantes con HSC suelen tener más complicaciones durante el embarazo, parto y puerperio que las mujeres sanas. Estas complicaciones son más frecuentes y graves en la AD que en la HSC. 26Se plantea que en esta etapa se agrava la anemia, y aumenta la incidencia de CVOD y STA. Además, son más frecuentes las trombosis placentarias, infecciones, toxemia, abortos espontáneos y muerte materna. 27El feto producto de la concepción también puede resultar afectado pues tiene mayor riesgo de retardo del crecimiento intrauterino, prematuridad, muerte intrauterina y perinatal, relacionados con la hipoxemia. 28
Sin embargo, la evolución del embarazo en este estudio fue favorable, no hubo mortalidad materna ni perinatal. Estos resultados son similares a los hallados en un estudio en el IHI en los años 2003-200429 y difieren de los reportados en Santiago de Cuba en el año 2008. 30 Esto debe estar relacionado con la atención médica altamente calificada por un equipo multidisciplinario que reciben las embarazadas con HSC en la institución. De los 26 abortos identificados en este estudio, la mayoría fueron espontáneos, lo que coincide con la literatura, 27e indica la necesidad de perfeccionar el seguimiento de la gestante con HSC desde su captación en la Atención Primaria de Salud con el objetivo de disminuir la posibilidad de abortar.
Comparando el resultado de las variables hematológicas con las reflejadas en un estudio de 114 pacientes con HSC efectuado en el centro de atención a la drepanocitosis en el Hospital Henri Mondor de Francia, 30 encontramos que los valores de Hb en ambos sexos fueron ligeramente inferiores a los descritos. En ambos estudios, el sexo masculino presentó una cifra de Hb superior, similar a lo que ocurre en la población general. El conteo de reticulocitos fue inferior, para lo cual no se tiene una explicación, aunque pudiera estar relacionado con una diferencia en la metodología para su estudio. El conteo de leucocitos estuvo dentro de los valores de referencia de normalidad lo cual constituye un factor pronóstico favorable.17La trombocitosis, considerada un factor pronóstico adverso, no estuvo presente en los pacientes estudiados. La Hb fetal también fue superior, esto pudiera deberse a que hubo un grupo de pacientes tratados con hidroxiurea, que aumenta la HbF.
En todos los grupos etarios de esta investigación predominaron los valores normales de creatinina y ALT lo que presumiblemente indica funciones hepática y renal normales. Resultados similares se reportan en Francia. 31
No obstante, cabe señalar que tanto los pacientes que presentaron elevación de la ALT como de las cifras de creatinina, fueron mayores de 40 años. Estos últimos desarrollaron posteriormente IRC, lo que indica que en la HSC se presenta daño orgánico crónico aunque es menos frecuente y más tardío que en la AD.14
En esta investigación la probabilidad de sobrevida global fue alta (62,6 %a los 50 años). En otras series se reportan datos muy similares.32En los Estados Unidos se reporta una expectativa de vida promedio de 60 años para los hombres y de 68 para las mujeres.33El número de fallecidos y las causas de muerte en el estudio fueron similares a las que se comunican en la literatura. 12,33La causa más frecuente fue la IRC, seguida por AVE, insuficiencia cardiaca y tromboembolismo pulmonar. La edad de la muerte también fue similar en ambas investigaciones, la mayoría por encima de los 25 años de edad.
El aumento de la calidad y expectativa de vida de la HSC en Cuba es el resultado de la implementación de los Programas Nacionales de Atención Integral y Diagnóstico Prenatal de la Drepanocitosis, que permiten el cuidado sistemático de los enfermos por un equipo especializado multidisciplinario y el fácil acceso a los servicios de urgencia y hospitalización.
REFERENCIAS BIBLIOGRÁFICAS
1. Wang WC. Sickle Cell Anemia and Other Sickling Syndromes. En: Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Bertil G. Wintrobe´s Clinical Hematology, 12th ed. Lippincott Williams and Wilkins; 2009.p.1039-82 (edición digital).
2. Roberts I, Montalembert M. Sickle cell disease as a paradigm of immigration hematology. New challenges for hematologists in Europe. Haematologica. 2007 Jul;92(7):865-71.
3. Morella B. Darlisona M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull WHO 2008;86(6):480-7.
4. Colombo B, Martinez G. Haemoglobinvariants in Cuba. Haemoglobin.1985;(9):415-22.
5. Mukisi-Mukaza M, Saint Martin C, Etienne-Julan M, Donkerwolcke M, Burny ME, Burny F. Risk factors and impact of orthopaedic monitoring on the outcome of avascular necrosis of the femoral head in adults with sickle cell disease: 215 patients case study with control group. OrthopTraumatolSurg Res 2011;97(8):814-20.
6. Markham MJ, LottenbergZumberg MA. Role of phlebotomy in the management of hemoglobin SC disease: case report and review of the literature. Am J Hematol. 2003;73(2):121-5.
7. Dacie JV. The hemolytic anemia: Congenital and acquired. Part I. The congenital anemias.2nd ed. New York. Grum&Stratton; 1960.
8. Svarch E, Hernández-Ramírez P, Ballester-Santovenia JM. La drepanocitosis en Cuba. Rev Cubana HematolInmunolHemoter [revista en la Internet]. 2004 Ago [citado 2012 Oct 15]; 20(2): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892004000200009&lng=es.
9. Normas para el tratamiento de la drepanocitosis. Grupo Nacional de Hematología y Bancos de Sangre. La Habana: Instituto de Hematología e Inmunología, 2013. [acceso: 16 de julio de 2013].Disponible en: http://www.sld.cu/sitios/hematologia
10. Mackerel TD, Cohen HW, Billet HH. The older sickle cell patient. Am J Hematol. 2004;76(1):101-6.
11. Morris J, Dunn D, Beckford M, GrandisonY, Mason K, Higgs D. et al. The haematology of homozygous SCD after the age of 40 years. Br J Hematol. 1991;77(2):382-5.
12. Shurafa MS, Prasad AS, Ruccknagel DL, KanYW. Long survival in sickle cell anemia. Am J Hematol. 1982;(12)357-65.
13. Losada-Buchillón R, Bravo-Cortada I, Kenneth C, Capildeo K, Agramonte O, Silva J. Pacientes con drepanocitosis y edad avanzada en Trinidad y Tobago. Rev Cubana HematolInmunolHemoter [revista en la Internet]. 2006 Ago [citado 2012 Oct 15]; 22(2): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892006000200007&lng=es.
14. Steinberg MH. Sickle cell disease and associated hemoglobinopathies. In: Goldman L, Schafer AI, eds. Cecil Medicine. 24th ed. Philadelphia: Saunders Elsevier; 2011.
15. Powars DR, Hiti A, Ramicone J C, Chan L. Outcome in Hemoglobin SC Disease: A Four-Decade Observational Study of Clinical, Hematologic, and Genetic Factors. Am J Hematol. 2002;70(1):206-15.
16. Koumbourlis AC, Lee DJ, Lee A. PediatrPulmonol. Lung function and somatic growth in patients with hemoglobin SC sickle cell disease. PediatrPulmonol. 2008 Feb;43(2):175-8.
17. John BJ, Schnog B, Lard LR, Rojer RA, Fey PL, Muskiet AJ.et al. New concepts in assessing sickle cell disease severity. Am J Hematol .1998; 58:61-2.
18. Villares-Álvarez I, Ríos-Araújo BT, Fernández-Águila JD, Aroche-Quintana M, Fojaco- Colina Y. Manifestaciones oculares en la drepanocitosis. Rev Cubana Oftalmol. 2009; Dic ; 22(2):131-9.
19. Leveziel N, Lalloum F, Bastuji-Garin S, Binaghi M, Bachir D, Galacteros F, et al. Sickle-cell retinopathy: Retrospective study of 730 patients followed in a referral center. J FrOphtalmol. 2012;35(5):343-7.
20. Boyd JH, Macklin EA, Strunk RC. Asthma is associated with increased mortality in individuals with sickle cell anemia. Hematol. 2007; 92(1):1115-8.
21. Morris CLR. Asthma management: reinventing the wheel in sickle cell disease. J Hematol. 2009;84(1):234-41.
22. Joshua J F, DeBaun MR. Asthma and sickle cell disease: two distinct, diseases or part of the same process?. Am Soc Hematol.2009; 1:45-7.
23. Gutiérrez-Díaz AI, Arencibia-Núñez A, Ramón-Rodriguez LG, Svarch E, Jaime-Fagundo JC, Machín-García S, et al . La drepanocitosis y el asma bronquial. Rev Cubana HematolInmunolHemoter [revista en la Internet]. 2013 [acceso: 16 de julio de 2013].Sep; 29(3): 233-45. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892013000300003&lng=es .
24. Ballas SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, et al. Beyond the Definitions of the Phenotypic Complications of Sickle Cell Disease: An Update on Management. Scientific World Journal. 2012; 2012: 949535.
25. Gillam AW, Lee RS, Hsi ED, Brotman DJ. Acute splenic sequestration crisis resembling sepsis in an adult with hemoglobin SC disease. South Med J 2004;97(4):413-5.
26. Serjeant GR, Hambleton IR, Thame M. Fecundity and pregnancy outcome in a cohort with sickle cell-hemoglobin C disease followed from birth. BJOG 2005;112(9):1308-14.
27. Iglesias-Hernández R, Casacó-Vázquez I, Silva-Barrios E, Vázquez-Cedeño JL, Ortiz- Jiménez Y. Complicación de una preeclampsia grave en una paciente portadora de hemoglobinopatía SC. Rev cuba anestesiolreanim [revista en la Internet]. 2009 Ago [citado 2013 Jun 24] ; 8(2): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1726-67182009000200008&lng=es.
28. Rogers DT, Molokie R. Sickle cell disease in pregnancy. ObstetGynecolClin North Am. 2010;37(2):223-37.
29. Hernández-Padrón C, Agramonte-Llanes O, Roque-Frías R, Ávila-Cabrera O, Mesa- Cuervo JR, Rodríguez-LR. Anemia drepanocítica y embarazo: transfundir o no transfundir, esa es la decisión. Rev Cubana HematolInmunolHemoter [revista en la Internet]. 2006 Ago [citado 2013 Jun 24]; 22(2): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892006000200010&lng=es.
30. Toirac LAS, Blanco RG, Pascual LV, Plasencia AC, Ibarra MM, Losada GJ. Hemoglobinopatías de tipo S y embarazo. Resultados en la atención al perinato. MEDISAN 2011;15(1):1
31. Lee K, C. Pre HU, G. Merault, L. Keclard, F.Thoraval R, Bachir D.et al. Genetic and Hematological Studies in a Group of 114 Adult Patients With SC Sickle Cell Disease. Am J Hematol 1998;59(1):15-21.
32. Prabhakar H, Haywood C, Malokie R. Sickle cell disease in the United States: Looking back and forward at 100 years of progress in management and survival. Am J Hematol 2010;(85):346-53.
33. Platt OS, Brambilla DJ, Rosse WF. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med;2003:1639-44.
Recibido: Agosto 21, 2013.
Aceptado: Noviembre 25, 2013
CORRESPONDENCIA A:
Dr. Sergio Machín García. INSTITUTO DE HEMATOLOGÍA E INMUNOLOGÍA. Apartado 8070, La Habana, CP 10800, CUBA.
Tel (537) 643 8695, 8268. Fax (537) 644 2334. Email: rchematologia@infomed.sld.cu
