










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter vol.31 no.3 Ciudad de la Habana jul.-set. 2015
ARTÍCULO ORIGINAL
Morbilidad y mortalidad de la anemia drepanocítica: estudio observacional de 36 años
Morbidity and mortality of sickle cell anemia: thirty six years of observational study
Dr. Sergio Machín GarcíaI, Dra. Idalsys Álvarez MolinaI, Prof. Eva SvarchI, Dra. Andrea Menéndez VeitíaI, Dr. Carlos Hernández PadrónI, Dra. Oramis Sosa PalaciosII
I Instituto de Hematología e Inmunología, La Habana, Cuba.
II Hospital Pediátrico Universitario “William Soler”, La Habana, Cuba.
RESUMEN
Introducción: la anemia drepanocítica es la anemia hemolítica congénita más común del mundo. Entre el 5 y el 15 % de la población mundial es portadora de la hemoglobina S. En Cuba es del 3,08 %, lo que representa un problema de salud pública.
Objetivo: identificar la morbilidad y la mortalidad en la anemia drepanocítica en el Instituto de Hematología e Inmunología (IHI).
Métodos: fueron estudiados todos los enfermos con seguimiento de al menos dos años en el IHI entre enero de 1973 y diciembre de 2009.
Resultados: se incluyeron 411 pacientes, de ellos, 215 mujeres. El seguimiento medio fue de 17,8 ± 9,8 años. El promedio anual de crisis vasoclusivas dolorosas, ingresos hospitalarios e infecciones fue de 1,8; 1,7; y 1,2, respectivamente. Las crisis vasoclusivas dolorosas, el síndrome torácico agudo y las crisis hepáticas fueron las manifestaciones clínicas más frecuentes, con ligero predominio en las mujeres, pero sin significación estadística en los dos últimos. El número de hospitalizaciones y consultas médicas fue mayor en los grupos de edades extremas. Los valores hematológicos y bioquímicos estaban dentro de los rangos esperados para la enfermedad, excepto los leucocitos; no hubo diferencias entre los sexos. Se diagnosticaron 132 embarazos en 86 pacientes. No hubo muertes maternas y sólo 5 muertes perinatales. La supervivencia global fue de 55 años. Las causas más frecuentes de muerte fueron las complicaciones hepáticas, accidentes vasculares encefálicos y la insuficiencia cardíaca.
Conclusiones: los pacientes fueron similares a lo descrito en la literatura, clínica y hematológicamente, excepto la frecuencia de complicaciones hepáticas que fue mayor. La probabilidad de supervivencia fue alta. Este estudio confirma la eficacia de los programas nacionales implementados en Cuba en el diagnóstico, seguimiento y tratamiento de la enfermedad.
Palabras clave: morbilidad, mortalidad, anemia drepanocítica.
ABSTRACT
Introduction: Sickle cell anemia (SS) is the most common congenital hemolytic anemia worldwide. The sickle cell trait (AS) comprises a range between 5 to 15% of the world population. In Cuba it is 3, 08 %, which represents a public health problem.
Objective: Identify morbidity and mortality in sickle cell anemia at the Institute of Hematology and Immunology (IHI).
Methods: All patients followed at least two years at the IHI between January 1973 and December 2009 were studied.
Results: 411 patients with sickle cell anemia were studied (January, 1973 to December, 2009), 215 female. Mean follow-up was 17.8 ± 9.8 years. The average annual vasoocclusive painful crises, hospital admissions and infection were 1.8; 1.7; and 1.2, respectively. Vasoocclusive painful crises, acute chest syndrome and hepatic crises were the most frequent clinical manifestations, with slight predominance in females, but not statistically significant in the last two. The number of hospitalizations and medical consultations was higher in extreme age groups. The hematological and biochemical values were within expected ranges for the disease except leukocytes; there were no differences between sex. One hundred thirty two pregnancies in 86 patients were diagnosed. There were no maternal deaths and only 5 perinatal deaths. Overall survival was 55 years. The most frequent causes of death were liver complications, strokes and heart failure.
Conclusions: Patients were similar clinical and hematologically as described in the literature, except the frequency of liver complications which was higher. The probability of survival was high. This study confirms the effectiveness of national programs implemented in Cuba in the diagnosis, monitoring and treatment of the disease.
Keywords: morbidity, mortality, sickle cell anemia.
INTRODUCCION
La anemia drepanocítica (AD) es la anemia hemolítica congénita más frecuente en el mundo1-3; donde la frecuencia del estado de portador AS abarca un rango que oscila entre el 5 y el 15 % 3. Cerca de 70 millones de personas padecen de AD y el 86 % de ellas se localizan en África Ecuatorial 4. También existe en el Medio Oriente, sur de Italia, norte de Grecia, sur de Turquía, provincias occidentales de Arabia Saudita y la India. Fue traída a Estados Unidos, América Central, el Caribe y algunos países de América del Sur por el comercio de esclavos 4.
La crisis vasoclusiva dolorosa (CVOD) representa el 90 % de los ingresos en sala de emergencia u hospitalización 5,6. En los adultos e incluso en los niños, la frecuencia e intensidad del dolor pueden ser subestimadas, ya que en varias ocasiones no acuden al hospital y solo lo hacen cuando es muy intenso 7. En estudios realizados se ha observado que el 25 - 40 % de los lactantes y niños de corta edad con AD sufren cuando menos un episodio de dactilitis 8, y los pacientes de mayor edad presentan episodios repetidos de dolor abdominal o músculo-esquelético 7.
El síndrome torácico agudo (STA) es la segunda causa de hospitalización y la primera de muerte en pacientes con AD. La etiología más frecuente en el niño es la infección y en el adulto, el infarto 9.
El accidente vascular encefálico (AVE) se debe a la oclusión de una arteria o a hemorragia3. Es más frecuente en la primera década de la vida; en el niño, la causa más frecuente es el infarto cerebral 10.
En la actualidad se plantea que pueden existir dos subfenotipos clínicos en la drepanocitosis. Uno caracterizado por hemólisis intravascular que produce vasculopatía sistémica por disminución del óxido nítrico, y clínicamente, por presentar úlceras maleolares, priapismo, hipertensión pulmonar, litiasis vesicular y AVE. En estos pacientes predominan la anemia y valores más elevados de reticulocitos, deshidrogenasa láctica, bilirrubina indirecta y hemoglobina (Hb) plasmática 3,11. En el segundo fenotipo predomina la oclusión vascular y se caracteriza por presentar CVOD, STA y osteonecrosis. Las cifras de Hb son mayores mientras que los reticulocitos, la deshidrogenasa láctica, la bilirrubina indirecta y la Hb plasmática tienen valores menos elevados 11.
El embarazo se asocia a la aparición de complicaciones de la enfermedad y puede poner a las mujeres en un riesgo adicional de complicaciones obstétricas. Históricamente, la mortalidad perinatal se ha informado tan alta como el 53 % para los bebés nacidos de mujeres con AD, pero la tasa se ha reducido drásticamente en las últimas tres décadas debido a una estricta atención hematológica, obstétrica y neonatal 11-13.
En 1973, la supervivencia media estimada de los pacientes con drepanocitosis fue de 14,2 años 14. Ya en el 1994 el promedio de sobrevida en el adulto era de 42 años en hombres y de 48 años en las mujeres 15. En el 2004, en el Instituto de Hematología e Inmunología (IHI) fue de 53 años 16.
En Cuba, el 3,08 % de la población es portadora de la hemoglobina S, por lo que representa un problema de salud pública y desde 1986 existe un Programa Nacional de Atención Integral de la Drepanocitosis 17.
El objetivo de esta investigación fue identificar la morbilidad y mortalidad en la AD en pacientes seguidos en el IHI durante un período de 36 años.
MÉTODOS
Se realizó un estudio observacional analítico, longitudinal y prospectivo. La población (N) estuvo conformada por 633 pacientes con drepanocitosis atendidos en el IHI en el período comprendido entre enero de 1973 y diciembre de 2009. La muestra (n) quedó constituida por 411 pacientes que cumplieron con los siguientes criterios de inclusión: diagnóstico confirmado de AD y seguimiento por más de 2 años en consulta.
La información de las variables objeto de estudio se obtuvo de las historias clínicas mediante una planilla confeccionada al efecto. En los casos de fallecimiento se revisó el certificado de defunción y el resultado del estudio anatomopatológico, siempre que estuvieron disponibles.
El proyecto de esta investigación fue aprobado por el Consejo Científico y el Comité de Ética de la institución. Dado que el estudio se realizó a partir del análisis de la historia clínica, no fue necesario el consentimiento de los pacientes para participar en la investigación. Se respetaron los criterios de confidencialidad y los datos obtenidos se emplearon para beneficio científico de interés institucional y de los pacientes.
Los exámenes complementarios se realizaron en condiciones basales del paciente, sin crisis ni transfusiones en los 3 meses previos a los exámenes. La hemoglobina se realizó por el método de la cianometahemoglobina y el recuento de leucocitos y plaquetas se realizaron en la cámara de Neubauer hasta el año 2008, que comenzó a utilizarse el equipo Mindray BC-3200. Para el estudio de hemoglobina fetal (Hb F) y los reticulocitos se emplearon los métodos recomendados en la literatura 18.
Se elaboró una base de datos en el programa SPSS 11.5.1, la cual fue procesada y analizada por medio de estadísticas descriptivas e inferencial. Desde el punto de vista descriptivo se calcularon frecuencias absolutas y porcentajes para las variables cualitativas; y la media y la desviación estándar para las cuantitativas. Se calculó la media y desviación estándar de ingresos y consultas por edades, del tiempo de seguimiento en años, del tamaño del bazo y el hígado por edades y de las variables hematológicas y bioquímicas por sexo. En las variables obstétricas se estimó el promedio de embarazos y otros indicadores cualitativos como son:
- Parto por embarazadas = Total de partos realizados X 100
Total de embarazadas
- Interrupciones por embarazadas = Total de interrupciones realizadas X 100
Total de embarazadas
Para el análisis de las variables obstétricas se excluyó del análisis al sexo masculino (n = 196), así como del sexo femenino a las mujeres fuera del rango de edad fértil (n = 69). Para el análisis de la frecuencia de priapismo se excluyeron las mujeres (n = 215).
Se aplicó análisis inferencial dado por pruebas de hipótesis de correlación lineal de Pearson (r) para identificar la relación entre las variables dependientes esplenomegalia y hepatomegalia con la edad (variable independiente); y análisis de varianza de una vía para identificar diferencias en las variaciones entre grupos (sexo,) de los resultados de pruebas hematológicas y bioquímicas (variable dependiente)
En el análisis inferencial se empleó un nivel de significación prefijado (p ≤ 0.05), y se consideró significativo cuando el valor probabilístico del error obtenido a partir de los datos fue menor que el p prefijado.
Se analizó la probabilidad de supervivencia por el método de Kaplan Meier.
RESULTADOS
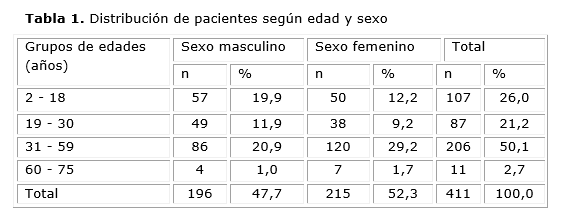
La distribución por grupos de edades y sexo se muestra en la tabla 1.
El tiempo medio de seguimiento fue de 17,8 ± 8 años.
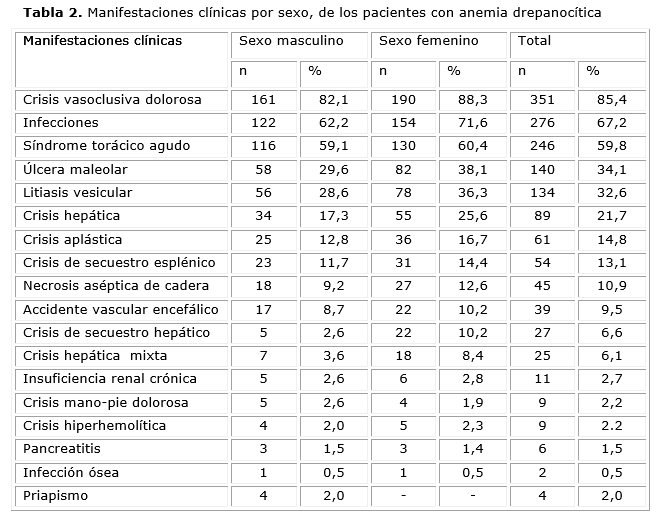
Las manifestaciones clínicas distribuidas por sexo se describen en la tabla 2.
El promedio anual de asistencia a consulta y el número de hospitalizaciones por año fue de 3,1 ± 2,1 y 1,7 ± 1,4 , respectivamente, con predominio en los extremos de la vida: 2 a 8 años (3,9 y 2,1) y más de 60 años (3,5 y 1,0).
En 309 pacientes se observó hepatomegalia (75,2 %) con una media de 1.8 cm, y la esplenomegalia se manifestó solo en 72 pacientes (17,5 %) con un promedio de 0.5 cm. En el grupo de 2 a 18 años la esplenomegalia alcanzó 1.2 cm en promedio, y fue menor en los restantes grupos de edades, al igual que la hepatomegalia. Al correlacionar el tamaño del bazo y el hígado con la edad se observaron variaciones (p <0.05) en sentido negativo: a mayor edad menor tamaño, más significativas en el bazo.
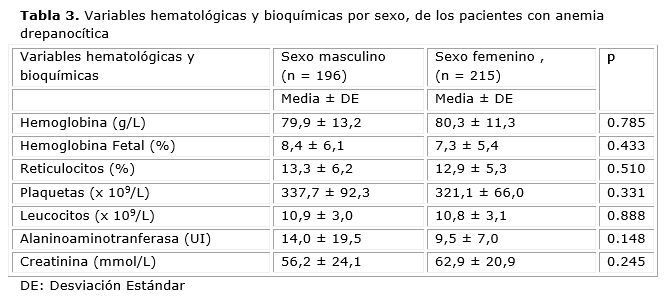
Los valores de la Hb, reticulocitos, plaquetas, leucocitos y Hb F se muestran en la tabla 3. La determinación de creatinina y ALAT no se realizó en todos los pacientes pero los valores estuvieron dentro del rango de la normalidad. No se evidenciaron diferencias significativas entre los sexos en el comportamiento de las variables hematológicas y bioquímicas (p >0.05).
En 27 (6,6 %) pacientes se recogió el diagnóstico de aloinmunización, con predominio de anticuerpos contra antígenos del sistema Rh.
En 146 mujeres en edad fértil hubo 132 embarazos; en 86 (58,9 %) se logró al menos un embarazo. Hubo un total de 81 partos (61,4 %), con 76 recién nacidos vivos (93,8 %). En el 38,6 % (n =,51) se interrumpió el embarazo; 25,5 % de ellos provocados y 19,6 % espontáneos. No hubo mortalidad materna y solo 5 muertes perinatales.
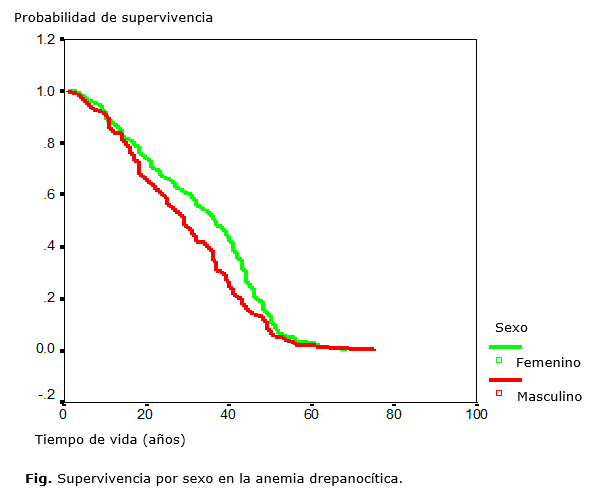
La probabilidad de supervivencia global de los pacientes fue de 55 años y no hubo diferencias de la supervivencia entre los sexos (figura).
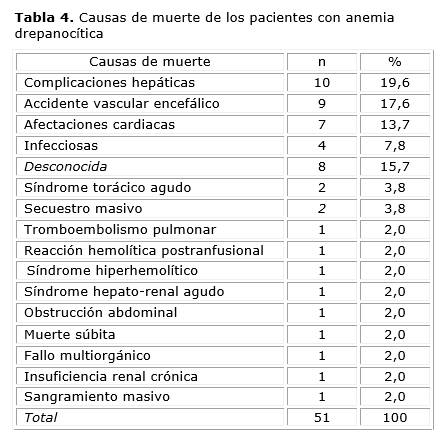
Las complicaciones hepáticas fueron la causa más frecuente de muerte (n,=,10; 19,6 %), seguidas por los AVE y las complicaciones cardiacas (tabla 4).
DISCUSIÓN
En este estudio hubo predominio del sexo femenino, contrario a lo esperado por el carácter autosómico de la enfermedad, por lo que no deben existir diferencias entre los sexos. La causa de este hallazgo no está clara pero posiblemente se relacione con la mayor estabilidad de las mujeres en consulta y que todas las embarazadas con drepanocitosis en la ciudad y las provincias vecinas son remitidas al IHI y gran parte de ellas continúan el seguimiento en la institución.
La mayor frecuencia de enfermos se localizó en la etapa adulta al momento del estudio, lo cual se justifica por las características de este que incluyó fundamentalmente adultos con un tiempo prolongado de seguimiento. Además, la implementación del Programa Nacional de Atención Integral de la Drepanocitosis dirigido a la prevención, control prenatal, tratamiento protocolizado y educación de los enfermos y familiares, ha logrado disminuir de forma importante la frecuencia de nuevos diagnósticos y la mortalidad de los niños y los adultos jóvenes.
La CVOD es el evento más frecuente en la AD 1,5,6,19, como se demostró en este estudio.
Las infecciones bacterianas son muy frecuentes en pacientes con AD. La pérdida temprana de la función esplénica los hace muy sensibles a las infecciones bacterianas. En el niño pequeño predomina la infección por neumococos y fue la primera causa de hospitalización y muerte en la primera infancia hasta la introducción de la penicilina oral profiláctica 20 y la vacuna contra el Streptococcus pneumoniae. En el niño mayor y en los adultos aparecen infecciones por microorganismos entéricos gram negativos 1. En el estudio se observó un promedio anual de casi dos infecciones por año, aunque solo 4 pacientes murieron por esta causa, lo cual se corresponde con la aplicación cotidiana e inmediata de medidas profilácticas y terapéuticas para la prevención y tratamiento de las infecciones.
El STA fue la manifestación clínica más frecuente después de las CVOD en esta investigación (59,8 %), superior a lo descrito en otros estudios 21,22. En la literatura se señala que la mortalidad durante el evento puede ser del 10 al 12 % y representa el 25 % de los fallecidos en la AD 21,23; sin embargo, en esta serie representó solo el 3,9 % de las causas de muerte. Esta baja mortalidad por STA puede ser el resultado de la educación a pacientes y familiares en el diagnóstico y la asistencia rápida a la sala de urgencias de la institución y la atención médica altamente calificada y precoz.
La frecuencia de úlcera maleolar (34 %) es comparable con lo reportado en países como Jamaica, pero muy superior a lo descrito en Estados Unidos 24,25. Llama la atención que en un estudio anterior realizado en la institución se encontró un número significativamente menor de esta manifestación clínica16, lo cual pudiera estar relacionado con el incremento del número de pacientes incluidos y al aumento del promedio de edad que es proporcional a la frecuencia de aparición de las úlceras 26.
El porcentaje de pacientes con litiasis vesicular fue similar al encontrado en países desarrollados pero menor que la que se comunica en Jamaica 27.
En la AD se describen lesiones hepáticas producidas por la falciformación crónica en los sinusoides, sobrecarga de hierro, hepatitis viral o una combinación de estos factores 28, aunque la literatura médica casi no menciona las complicaciones agudas 28,29. En este estudio, 148 (36 %) pacientes presentaron alguna manifestación hepática aguda, para lo que no se tiene una explicación clara. En otros estudios realizados en la institución no se ha encontrado una explicación lógica de este hallazgo 16,29.
El AVE se presenta entre el 6 y el 12 % de los pacientes con AD. En el grupo estudiado se observó un porcentaje semejante a lo publicado por otros autores 5,10,29.
La insuficiencia renal crónica (IRC) puede presentarse entre el 4 y el 20 % de los pacientes con AD, con una edad media de aparición de 23 años 30,31. En esta investigación, la IRC se presentó en una frecuencia menor a la reportada internacionalmente, pero muy inferior a la descrita en el estudio realizado en el IHI en 2004 16, lo que pudiera estar relacionado también con el incremento del número de pacientes incluidos, además de la detección temprana de microalbuminuria y el inicio de su tratamiento con los inhibidores de la enzima convertidora de la angiotensina 31,32.
La incidencia del resto de las manifestaciones clínicas fue similar a lo descrito por otros autores 33.
El promedio anual de consultas y hospitalizaciones fue mayor en las edades extremas. En la edad pediátrica se puede atribuir a que los niños ingresan más por eventos de la enfermedad más frecuentes en este grupo de edad, y por la responsabilidad de los padres en su atención médica. En el adulto mayor debe estar relacionado con el incremento de las manifestaciones clínicas secundarias a la disfunción orgánica crónica.
La esplenomegalia tuvo una correlación inversa con la edad. Se conoce que en el bazo hay un proceso crónico de oclusión de los sinusoides esplénicos que conduce a la fibrosis y a la autoesplenectomía 1,33. Alrededor de los 6 a 8 años de edad el órgano deja de ser palpable.
La hepatomegalia constituye un signo clínico prácticamente constante en la AD; casi siempre es consecuencia del proceso hemolítico crónico, de infecciones víricas por transfusiones y hemosiderosis 1. Es más frecuente en adultos; sin embargo, en este estudio se observó una disminución estadísticamente significativa con la edad, para lo cual no se cuenta con una explicación. La crisis de secuestro hepático es muy rara y menos severa que la crisis de secuestro esplénico del niño1, como se observó en este estudio.
Los valores de Hb, reticulocitos, plaquetas y Hb F fueron similares a lo descrito en otras series. Sin embargo, no se encontró leucocitosis, como se plantea en casi todos los estudios 1,5,33. La media de la ALAT y la creatinina estuvieron dentro de rangos normales. En general, las variables hematológicas y bioquímicas no experimentaron variaciones importantes según el sexo.
Las transfusiones tienen indicaciones precisas y no están exentas de riesgos, entre ellos, la reacción hemolítica postransfusional por la aparición de anticuerpos contra antígenos de los eritrocitos del donante, proceso denominado aloinmunización. El porcentaje de pacientes aloinmunizados fue mayor a lo comunicado en países que utilizan técnicas de biología molecular en el inmunofenotipaje de las donaciones y los pacientes 34,35, pero menor comparado con la mayoría del resto del mundo. En el centro, desde hace mucho tiempo, se realiza inmunofenotipaje serológico ABO y del sistema Rh en todos los pacientes politransfundidos, lo que ha permitido disminuir notablemente el índice de aloinmunización en la AD.
Aunque se conoce la mayor frecuencia de complicaciones y mortalidad durante la gestación 12, no se prohíbe un embarazo deseado a la mujer con AD. La evolución del embarazo en estas pacientes fue muy buena. No hubo mortalidad materna y la perinatal fue muy baja. Estos resultados son muy superiores a lo descrito en otros países 12,36 y es el fruto del trabajo conjunto de hematólogos, obstetras, intensivistas y neonatólogos.
La sobrevida global fue de 55 años, similar a la alcanzada en centros médicos de gran desarrollo en el mundo 15,33,37. No hubo diferencias significativas en la sobrevida por sexo.
El STA, la insuficiencia renal crónica, las infecciones y los AVE son las causas de muerte más frecuentes descritas en la literatura 33. Sin embargo, las complicaciones hepáticas fueron la primera causa de muerte en el presente estudio, para lo cual tampoco se tiene una explicación clara. Se recogió el diagnóstico de hepatitis B o C en 22 (5,3 %) de los enfermos, inferior a lo reportado en otros trabajos 27, lo que está en contra de la posible etiología viral clásica del fenómeno.
Los resultados de este estudio confirman la eficacia de los programas nacionales aplicados para el diagnóstico, seguimiento y tratamiento de la AD en Cuba, así como la importancia de la atención integral por grupos multidisciplinarios altamente calificados en el manejo de la enfermedad y sus complicaciones.
REFERENCIAS BIBLIOGRAFICAS
1. Wang WC. Sickle Cell Anemia and Other Sickling Syndromes. En: Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Bertil G. Wintrobe´s Clinical Hematology, 12th ed. Philadelphia: Lippincott Williams and Wilkins; 2009. p. 1039-82.
2. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010 May(1):104–10.
3. Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global Burden of Sickle Cell Anaemia in Children under. Five, 2010–2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013Jul;10(7)1-14.
4. Ware RE. Is Sickle Cell Anemia a Neglected Tropical Disease? PLoS Negl Trop Dis. 2013;7(5):1-4. doi: 10.1371/journal.pntd.0002120.
5. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010 Dec 11;376(9757):2018-31.
6. Smith WR. Pain in sickle cell disease: the future of acute treatment. Expert Rev Hematol. 2011;4(3):237–9.
7. Smith WR, Scherer M. Sickle-cell pain: advances in epidemiology and etiology. Hematology Am Soc Hematol Educ Program. 2010;2010:409-15.
8. Almeida A, Roberts I. Bone involvement in sickle cell disease. Br J Haematol. 2005 May;129(4):482-90.
9. Scott TM. How I treat acute chest syndrome in children with sickle cell disease. Blood. 2011 Mar;117(15):5297-305.
10. Webb J, Kwiatkowski J. Stroke in patients with sickle cell disease. Expert Rev Hematol. 2013 Jun;6(3):301-16.
11. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;2:37-47.
12. Costa VM, Viana MB, Aguiar RA. Pregnancy in patients with sickle cell disease: maternal and perinatal outcomes. J Matern Fetal Neonatal Med. 2014 Jun:1-5.
13. Hassell KL. Pregnancy and sickle cell disease. Hematol Oncol Clin North Am. 2005 Oct;19(5):903-16.
14. Diggs LW. Anatomic lesions in sickle disease. In: Abramson H, Bertles JF, Wethers D. Sickle cell disease: diagnosis, management, education, and research. St. Louis: Mosby; 1973.p.189–229.
15. Platt OS, Brambilla BJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994 Jun;330(23):1639-44.
16. Machín García S, Guerra Alfonso T, Svarch E, Espinosa Martínez E, Mesa Cuervo JR, Dorticós Balea E, et al. Morbiletalidad en pacientes adultos con drepanocitosis. Rev Cubana Hematol Inmunol Hemoter [revista en la Internet]. 2004 Ago [citado 2014 Sept 22];20(2):
Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892004000200004&lng=es.
17. Svarch E, Marcheco Teruel B, Machín García S, Menéndez Veitía A, Nordet Carrera I, Arencibia Núñez A, et al. La drepanocitosis en Cuba. Estudio en niños. Rev Cubana Hematol Inmunol Hemoter. 2011;27(1):51-67.
18. Betke K, Marti H, Schicht I. Stimulation of small percentages of foetal hemoglobin. Nature. 1959;184:1877-81.
19. Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: Frequency, etiology and prognostic significance. Am J Hematol. 2005 May;79(1):17-25.
20. Hirst C, Owusu-Ofori S. Prophylactic antibiotics for preventing neumococcal infection in children with sickle cell disease. Cochrane Database Syst Rev. 2012 Sep 12;9:CD003427. doi: 10.1002/14651858.CD003427.pub2.
21. Desai PC, Ataga KI. The acute chest syndrome of sickle cell disease. Expert Opin Pharmacother. 2013 Jun;14(8):991-9.
22. Abbas HA, Kahale M, Hosn MA, Inati A. A review of acute chest syndrome in pediatric sickle cell disease. Pediatr Ann. 2013 Mar;42(3):115-20.
23. Perronne V, Roberts-Harewood M, Bachir D, Roudot-Thoraval F, Delerod JM, Thuret I, et al. Patterns of mortality in sickle cell disease in adults in France and England. Hematol J. 2002;3(1):56-60.
24. Cumming V, King L, Fraser R, Serjeant G, Reid M. Venous incompetence, poverty and lactate dehydrogenase in Jamaica are important predictors of leg ulceration in sickle cell anaemia. Br J Haematol 2008 Jul;142(1):119-25.
25. Delaney KM, Axelrod KC, Buscetta A, Hassell KL, Adams-Graves PE, Seamon C, et al. Leg ulcers in sickle cell disease: current patterns and practices. Hemoglobin. 2013;37(4):325-32.
26. Minniti CP, Eckman J, Sebastiani P, Steinberg MH, Ballas SK. Leg ulcers in sickle cell disease. Am J Hematol. 2010 Oct;85(10):831-5.
27. Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol. 2010 Jun;8(6):483-9.
28. Koskinas J, Manesis EK, Zacharakis GH, Galiatsatos N,Sevastos N, Archimandritis AJ. Liver involvement in acute vaso-occlusive crisis of sickle cell disease: prevalence and predisposing factors. Scand J Gastroenterol. 2007;42:499-507
29. Svarch E, Hernández Ramírez P, Ballester Santovenia JM. La drepanocitosis en Cuba. Rev Cubana Hematol Inmunol Hemoter [en línea] 2004 Ago [citado 2012 Sept 26] ;20(2):.
Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892004000200009&lng=es.
30. Núñez-Quintana A, Hondal-Álvarez N, Ayllón-Valdés L. Alteraciones renales en la drepanocitosis. Rev Cubana Hematol Inmunol Hemoter. 2011;27(2):168-78.
31. Sharpe CC, Thein SL. How I treat renal complications in sickle cell disease. Blood. 2014 Jun;123(24):3720-6.
32. Ataga KI, Derebail VK, Archer DR. The glomerulopathy of sickle cell disease. Am J Hematol. 2014 Sep;89(9):907-14.
33. Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med. 2013 Oct;3(10):a011783.
34. Mijovic A, Perera IG, Thein SL. Red blood cell alloimmunization in sickle cell disease-prevalence and trends: a single-center cross-sectional study from United Kingdom. Transfusion. 2013 Dec;53(12):3279-80.
35. Matteocci A, Pierelli L. Red blood cell alloimmunization in sickle cell disease and in thalassaemia: current status, future perspectives and potential role of molecular typing. Vox Sang. 2014 Apr;106(3):197-208.
36. Resende Cardoso PS, Lopes Pessoa de Aguiar RA , Viana MB. Clinical complications in pregnant women with sickle cell disease: prospective study of factors predicting maternal death or near miss. Rev Bras Hematol Hemoter. 2014 Jul-Aug;36(4):256-63.
37. Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010 Apr;115(17):3447-52.
Recibido: octubre 10, 2014.
Aceptado: enero 19, 2015.
Dr. Sergio Machín García. Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, Cuba. Tel (537) 643 8695, 8268. Email: rchematologia@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}