










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter vol.32 no.1 Ciudad de la Habana ene.-mar. 2016
PRESENTACIÓN DE CASO
Síndrome de hiper IgE autosómico dominante
Autosomal dominant hyper IgE síndrome
Daily Pino Blanco, Imilla Casado Hernández, Consuelo Macías Abraham, Lázaro O. del Valle Pérez, Odalis M. de la Guardia Peña, Miriam Sánchez Segura
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
El síndrome hiper IgE autosómico dominante es una inmunodeficiencia primaria poco frecuente que se caracteriza por niveles elevados de IgE, dermatitis eccematoide, infecciones recurrentes de piel y pulmón, y formación de abscesos con escasos signos inflamatorios. También se presentan alteraciones dentarias, esqueléticas y del tejido conjuntivo. Es causado por mutaciones dominantes del gen que codifica la proteína transductora de señal y activadora de la transcripción 3 (STAT3). Esta mutación condiciona un déficit en la generación de células Th17 a partir de células T CD4+, que explica la susceptibilidad de estos pacientes a infecciones por S aureus y C albi cans. Se presenta una adolescente con puente nasal amplio, paladar ojival, hiperlaxitud, fracturas patológicas, escoliosis y retraso en la caída de la dentadura primaria, rash eccematoso desde el período neonatal, infecciones cutáneas, óticas, pulmonares y candidiasis mucocutánea. Se detectan niveles elevados de IgE sérica y eosinofilia. Ha sido tratada con antimicrobianos y tópicos, con seguimiento de más de 10 años. Este síndrome es una entidad infrecuente, de causa genética, que requiere alto grado de sospecha y del manejo precoz de las infecciones.
Palabras clave: síndrome de hiperIgE autosómico dominante, enfermedad de Job, inmunodeficiencia primaria.
ABSTRACT
Autosomal dominant hyper IgE syndrome is a rare primary immunodeficiency characterized by elevated levels of IgE, eczematoid dermatitis, recurrent infections of skin and lung and abscess formation with few inflammatory signs. Dental, skeletal and connective tissue disorders are also present. It is caused by dominant mutations of the gene encoding the protein signal transducer and activator of transcription 3 (STAT3) . This mutation deficit conditions in generating Th17 cells from CD4 + T cells which explains the special susceptibility of these patients to infection by S. aureus and Candida albicans. A teenager patient is presented, broad nasal bridge, arched palate, hypermobility, pathological fractures, scoliosis and fall of primary teeth delayed, eczematous rash from neonatal lung, skin infections, ear and mucocutáneous candidiasis. High levels of Ig E serum and eosinophilia were detected. The patient was treated with antibiotics and topical, tracking over 10 years. Conclusions: This syndrome is a rare condition, genetic causes require high degree of suspicion and early management of infections.
Keywords: autosomal dominant hyper IgE syndrome, Job disease, primary immunodeficiency.
INTRODUCCIÓN
Las inmunodeficiencias primarias (IDP) son enfermedades en las que un defecto genético produce una alteración del sistema inmune que se traduce en susceptibilidad a infecciones, autoinmunidad, alergias y predisposición al desarrollo de tumores malignos.1 Existen más de 150 enfermedades catalogadas como tales y en la mayoría se conoce el defecto molecular que las ocasiona, aunque se siguen describiendo nuevos genes cuyas mutaciones originan IDP y nuevos fenotipos clínicos.
Aunque vistas en forma aislada se podrían considerar poco frecuentes, las IDP tienen una incidencia de 1 en 8 000 a 10 000 niños nacidos vivos, y una prevalencia de 1/250 hasta 1/500 (excluyendo el déficit de IgA, que es asintomático en la mayoría de los casos), comparable con la prevalencia de 1/700 para la diabetes tipo I y 1/1 000 para la esclerosis múltiple.1 Diferentes alteraciones cutáneas pueden ser su manifestación inicial.
Entre las IDP con compromiso cutáneo, se encuentra el síndrome de hiper IgE (SHIE), también conocido como síndrome de Job, nombrado así en1966 cuando fueron descritos los casos de dos niñas con abscesos recurrentes «fríos» por estafilococos, neumonía y un rash eccematoide de aparición en el periodo neonatal, que les recordaron un pasaje bíblico en el Libro de Job. Años más tarde (1972) Buckley y col comunicaron por primera vez la existencia en estos pacientes de niveles séricos de IgE excepcionalmente elevados, con concentraciones del resto de las inmunoglobulinas dentro de la normalidad.2
En 1999 se estableció definitivamente la forma de transmisión del SHIE clásico, a través de una herencia autosómica dominante con penetrancia variable (SHIE-AD). Años más tarde se identificó una mutación homocigota del gen de la tirosina cinasa 2 (Tyk2) como causa molecular del SHIE autosómico recesivo (SHIE-AR); recientemente, se describió la mutación de DOCK8 (dedicator of cytokinesis 8) como otra forma de transmisión de SHIE-AR y otro patrón AR cuya patogenia es desconocida.2-5
El SHIE se considera un desorden disregulatorio raro y complejo, caracterizado por hiperglobulinemia E, eosinofilia, abscesos cutáneos, dermatitis eccematoide crónica, candidiasis mucocutánea crónica e infecciones pulmonares recidivantes que contribuyen al desarrollo de neumatoceles y bronquiectasias, cuya herencia puede ser autosómica recesiva o dominante.3,4 Se clasifica en dos tipos: I, SHIE-AD, en el cual los pacientes se presentan con anormalidades en distintos sistemas incluido el inmunológico, tejido conectivo, esquelético y vascular entre otros; y el tipo II, SHIE-AR, que también afecta al sistema inmune, manifestado como IgE elevada, infecciones recurrentes de piel y pulmón, susceptibilidad a infecciones virales como Molluscum contagiosum, y compromiso del sistema nervios central, pero sin las alteraciones musculoesqueléticas.5
Los genes involucrados en el SHIE son: STAT3 (signal transducer and activator of transcription 3), DOCK8 y Tyk2. Independientemente del defecto molecular, los pacientes con SHIE comparten rasgos comunes pero diferente compromiso clínico e inmunológico; en aproximadamente la mitad de los casos se desconoce el defecto molecular.6
Aproximadamente el 25 % de los pacientes con SHIE presentan mutación en STAT3. En ellos se observan las características inmunológicas antes descritas con un variado rango de manifestaciones somáticas no inmunológicas, que incluyen: facies característica, que se evidencia completamente luego de la adolescencia con frente y barbilla prominentes, base nasal ancha, craneosinostosis, retención de dientes primarios, paladar ojival, hiperlaxitud, escoliosis y osteoporosis que lleva a fracturas con traumatismos mínimos. Por otro lado, estos pacientes tienen riesgo elevado de desarrollar malignidad, particularmente linfoma no hodgkiniano y aneurismas arteriales.6
Aquellos que presentan mutaciones en DOCK8 desarrollan una respuesta inmune defectuosa. En ellos, los linfocitos T (CD3+CD4+ y CD3 +CD8+) están disminuidos, presentan deterioro proliferativo luego de la estimulación in vitro y alteración en la producción de citocinas antivirales. A diferencia del SHIE-AD, no presentan anormalidades esqueléticas y dentarias, si bien tienen incremento de complicaciones autoinmunes. Las mutaciones en DOCK8 generan un fenotipo de SHIE con menor compromiso pulmonar pero mayor expresión de síntomas cutáneos y aumento de la susceptibilidad a infecciones virales. Las deficiencias en Tyk2 presentan en especial IgE en valores menos elevados e incluso normales, con un fenotipo clínico muy variable.6
Desde el punto de vista inmunológico los pacientes presentan una disfunción en la respuesta inflamatoria, con acción deficiente frente a agentes infecciosos y una reparación tisular aberrante asociada a incapacidad para elaborar una respuesta Th17 efectiva.4,5
Se describe el caso de una adolescente con SHIE-AD; se presentan las características clínicas, exámenes de laboratorio y aspectos fisiopatológicos, ante la novedad diagnóstica que representa este caso que, hasta donde se conoce, es el primero diagnosticado en edades pediátricas.
PRESENTACIÓN DEL CASO
Paciente femenina de 13 años de edad, mestiza, con antecedentes de piodermitis y eccema severo en piel desde los 15 días de nacida. Desde ese momento comenzó con neumopatías a repetición, que requirieron ingreso hospitalario en reiteradas ocasiones que se han mantenido durante toda su vida. Desde esa misma etapa comenzó con otitis media aguda recurrente y candidiasis oral/vaginal refractaria a la terapia antimicótica. Durante un largo período presentó en tórax una lesión papulomatosa con vesículas, sugestiva de una histiocitosis de células grandes, descartada por biopsia. Además, había presentado múltiples abscesos cutáneos en piel resistentes al tratamiento, intervenidos quirúrgicamente, y cuyo cultivo en todos los casos demostró la presencia de Staphilococcus aureus.
Durante la evolución de la enfermedad realizó ciclos de tratamiento con antimicóticos por vía oral y tópica, antihistamínicos y antibióticos de varias generaciones, en altas dosis, incluidas las sulfas, sin evidenciarse mejoría clínica.
Ante la no resolución de los cuadros infecciosos y la tórpida evolución, a la edad de 12 años acudió a la consulta de Inmunología del Instituto de Hematología e Inmunología. La valoración de la historia de la enfermedad y los signos encontrados en el examen físico, hicieron sospechar del diagnóstico del SHIE. Se le indicaron exámenes de laboratorio, imagenológicos y microbiológicos para corroborar dicho diagnóstico.
Al examen físico se encontró: facies característica (puente nasal hundido, base nasal ancha, protrusión de la frente y leve prognatismo), aumento de la distancia intercantos externos y piel engrosada con poros dilatados; retención de la dentición primaria, paladar ojival, depresión central lingual, con fisuras y arrugas de la mucosa oral. Como manifestaciones musculoesqueléticas principales: sifoescoliosis, fracturas patológicas e hiperlaxitud articular (figura 1).

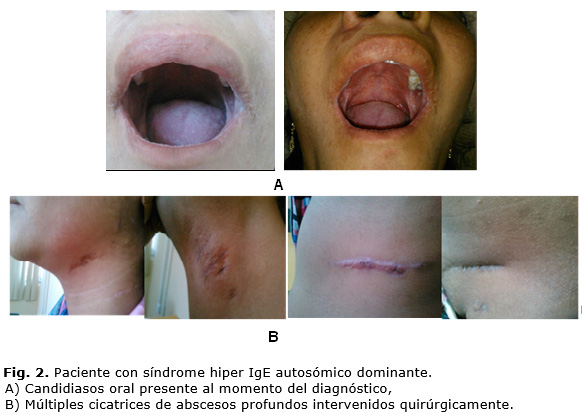
Se encontraron lesiones eccemáticas en piel en la región del cuello, miembros inferiores, superiores y abdomen, que se exacerbaban en algunos períodos pero no desaparecían; múltiples cicatrices en el cuello, región axilar, abdomen, espalda y ambos miembros, causadas por las intervenciones quirúrgicas de los abscesos profundos y la presencia de una candidiasis oral que requirió tratamiento local y sistémico (figura 2) .
Las cifras de hemoglobina y leucocitos se encontraban dentro de parámetros normales. En el estudio citomorfológico se observó eosinofilia moderada, con valores absolutos de eosinófilos de 830 X 109 (VN: hasta 350 X 109); nivel de IgE mayor de 200 UI/mL (VN: 150 UI/mL).
Se constató ligero retardo de la fagocitosis, expresado a los 15 y 60 min, T15: 85,2 % (VN: 22,99 a 53,95 %), T60: 54,97 % (VN: 6,63 a 28,43 %); y valores discretamente bajos del componente 3 del complemento 0,89g/L (VN: 0,90-1,70g/L). Las pruebas de anticuerpos anticitoplasma de neutrófilos (ANCA) y de alergia a diferentes alergenos, resultaron negativas. La cuantificación de inmunoglobulinas resultó dentro de la normalidad, comparada con valores de referencia según la edad. El estudio celular por citometría de flujo reveló la disminución de los valores de células CD3-CD56+ 5,53 % (VN: 6-27 %)
El exudado nasofaríngeo resultó positivo para S aureus; así como el estudio microbiológico de las lesiones en piel, que evidenció infección por Candida albicans.
Se interconsultó con otras especialidades como dermatología, endocrinología y alergia, cuyos criterios fueron valiosos para el diagnóstico diferencial.
Según el cuadro clínico, los exámenes complementarios, los criterios de Grimbacher (mayor de 40 puntos) (tabla) y lo debatido con otros especialistas, se concluyó que el diagnóstico de la paciente era un SHIE, posiblemente heredado de manera AD, a pesar de no contar con el apoyo de estudios moleculares y genéticos.
Las pautas terapéuticas se fundamentaron en la orientación de medidas profilácticas para el cuidado de la piel, mucosas y de las vías respiratorias. Así como en el tratamiento medicamentoso para la estimulación/modulación del sistema inmunológico y el control de los síntomas. Se le indicó factor de transferencia (extracto leucocitario de producción nacional, Hebertrans, SA) 1 unidad subcutánea semanal en ciclos de 8 semanas; gammaglobulina humana al 10 %, 5 bulbos en dosis fraccionadas, por vía intramuscular cada 15 días, 8 dosis; vitamina C, 2 g diarios vía oral; antihistamícos como la cimetidina, 2 tabletas por día; la ciproheptadina, 4 mg, 2 tabletas por día; y el ketotifeno, 1 mg, 2 tabletas diarias, en dependencia del predominio de los síntomas dermatológicos o respiratorios, respectivamente; varios ciclos de complejo vitamínico Truabin, tres veces por semana por 12 semanas, por vía intramuscular, a descansar 4 semanas; y un ciclo de terapia con aceite ozonizado, por vía tópica y Nistatina suspensión por vía oral, para el control de la candidiasis mucocutánea.
Durante el cumplimiento del tratamiento se evidenció una notable mejoría clínica; la paciente permaneció asintomática durante un largo período, en el que se comprobó disminución de las lesiones eccematosas y abscesos en piel, desaparición de los cuadros respiratorios y de las infecciones micóticas.
DISCUSIÓN
El caso clínico presentado cumple con las manifestaciones y la evolución del SHIE-AD.4,6
Por no disponer del estudio genético, el diagnóstico se basó en la clínica de acuerdo con los criterios del Instituto Nacional de Salud de los Estados Unidos (NIH), denominados Criterios de Grimbacher, que establecen el diagnóstico como altamente probable si reúne más de 40 puntos, posible entre 20 y 40 puntos e improbable con menos de 20 puntos. El caso presentado sumó más de 40 puntos en este sistema (tabla).
Los pacientes con estas mutaciones presentan características inmunológicas, tales como IgE mayor que 2 000 IU/mL (97 %), eccema moderado-severo (95 %), eosinofilia (90 %), abscesos (87 %), neumonías recurrentes (87 %), candidiasis mucocutánea (83 %), entre otras.5,7,8
También muestran un amplio rango de manifestaciones somáticas no inmunológicas presentes en el caso que se reporta.4,7-9
Una característica típica del SHIE-AD es la inflamación aberrante, que lleva a la producción de abscesos fríos en la piel con una respuesta inflamatoria exagerada, principalmente pulmonar, que provocaría la formación de neumatoceles. Esto se debe a que el STAT3 actúa como un activador de la señalización de diferentes receptores de citocinas, que incluyen IL-6, IL-10, IL-11, IL-17, IL-21, IL-22 e IL-23, entre otros. El hecho de que STAT3 participe, tanto en vías de citocinas pro como antinflamatorias, conduce a una desregulación que promueve o inhibe la inflamación según las circunstancias.4
Las mutaciones en STAT3 determinan una falla en la diferenciación Th17 y en la secreción de IL – 17.2,4 Se ha demostrado que una deficiencia en el linaje Th17 incrementa la susceptibilidad de la infección por S aureus, y los efectos protectores de IL-17 fueron apreciables en pacientes con SHIE, los cuales son susceptibles a infecciones recurrentes por este germen. Las mutaciones en STAT-3 resultan en una falla de los linfocitos T CD4+ para diferenciarse en células Th17. Se conoce que IL-6 e IL-23 son claves en el desarrollo de Th17 y activan STAT-3. La deleción de STAT3 se asocia, además, a la generación de osteoclastos, osteopenia de fenotipo osteoporótico y niveles más elevados de reabsorción ósea.2,10,11
Las células Th17 secretan también IL-22, la cual participa en la regulación positiva de distintas células para la producción de CCL20 y β defensinas, como por ejemplo, queratinocitos y células epiteliales pulmonares. Se ha demostrado en pacientes con forma AD, la reducción de la producción de esos péptidos antimicrobianos. Por otro lado, la anormal remodelación pulmonar hallada luego de infecciones o cirugías, se ha relacionado con la participación de STAT3 en la regulación de distintas metaloproteinasas de la matriz pulmonar.2,4,10 La disminución de la producción de defensinas en la piel y el pulmón puede explicar los abscesos en piel y pulmón característicos del SHIE.
Además, existe una alteración en la estimulación de la producción del factor antiestafilocócico del epitelio bronquial y los queratinocitos, lo que explicaría la restricción de las infecciones por S aureus a estos órganos.2 Estas neumopatías pueden ocurrir más tardíamente, particularmente en niños tratados con profilaxis antibiótica; sin embargo, en este caso aparecieron en etapas tempranas de su vida.12
En general, el S. aureus es la bacteria más frecuentemente aislada en las muestras microbiológicas de estos pacientes. Durante los episodios infecciosos también pueden aislarse: Streptococcus pneumoniae, Haemophylus influenza, enterobacterias, Pseudomona aeruginosa y gérmenes oportunistas como Pneumocystis jiroveci y Criptococcus, causantes de neumopatías e histoplasmosis. . Los pacientes con SHIE presentan, además, susceptibilidad a infecciones fúngicas que incluyen moniliasis oral y onicomicosis por C albicans. Las infecciones tienen tendencia a formar abscesos y pneumatoceles o quistes pulmonares, que pueden sobreinfectarse por Aspergillus fumigatus, Pseudomonas aeruginosa multirresistentes o Mycobacterium no-tuberculoso.2,4
Otro hallazgo es la presencia de dermatitis crónica, pruriginosa, evidente ya en las primeras semanas de vida. Sus características y distribución difieren de la dermatitis atópica, con mayor compromiso en la cara, axilas, ingles, periné y superficies de extensión; con poca base eritematosa. A diferencia de los pacientes con dermatitis atópica, en el SHIE suele faltar el antecedente familiar de atopia y las infecciones cutáneas no son solamente superficiales sino que afectan a tejido celular subcutáneo.7
El retraso en la caída de la dentición primaria, es otro rasgo clínico que puede aparecer y ayudar al diagnóstico, observable hasta en el 60 - 72 % de los pacientes. El retraso en la caída de los dientes primarios asociado a la erupción de los sucedáneos, puede determinar la presencia de una doble hilera de dientes.12
Las anomalías óseas y del tejido conjuntivo pueden presentarse a cualquier edad. Los pacientes pueden presentar fracturas con poco dolor y densidad ósea eventualmente normal. Sin embargo, algunas lesiones características, particularmente el aplastamiento de las vértebras lumbosacras, se encuentran casi exclusivamente en adultos o niños mayores y se asocian con osteoporosis pero no con anomalías del metabolismo fosfocálcico. Más del 75 % de los mayores de 11 años presentan escoliosis. También puede observarse hiperextensibilidad de las articulaciones.12 Se considera que el defecto de señalización de IL-11 sería la causa primordial de las malformaciones óseas y dentales.4
En el estudio inmunológico se destacan elevados valores de IgE, generalmente superiores a 2 000 IU/mL. Existe un porcentaje de recién nacidos con SHIE que pueden tener valores normales y hasta el 20 % de los adultos llegan a normalizar los valores. El aumento no es proporcional a la gravedad del cuadro y gran porcentaje está dado por IgE específica a S aureus y C albicans. Los niveles de IgG, IgA e IgM generalmente se encuentran dentro del rango normal.
Habitualmente el hemograma no muestra alteraciones, aunque se ha descrito neutropenia
relativa. Algunos pacientes muestran eosinofilias relativas de hasta 40 - 50 % y absolutas mayores a 2 desviaciones estándar.6 La producción del factor estimulador de colonias de granulocitos y monocitos se ha encontrado elevada en estos pacientes, lo que en parte explica la eosinofília.2,9,10
La mayoría de los autores reportan valores normales de linfocitos T, deficiente producción de anticuerpos específicos y disminución de los linfocitos B de memoria. Los hallazgos encontrados en el caso clínico que se presenta coinciden con otro grupo que refiere ligera disminución de la inmunidad celular.3,5,10,12
A diferencia de lo descrito en la enfermedad granulomatosa crónica, en este caso se comprobó un retardo en el índice opsonofagocítico. Las células Th17, comprometidas en el SHIE-AD, son importantes en el reclutamiento de neutrófilos y la adhesión celular, eventos claves en la inmunidad frente a patógenos. Por otra parte, el déficit en la producción de IL12 no favorece la activación de los macrófagos, con lo que se afecta la fagocitosis.2,5,10 Algunos autores refieren que existe deficiencia de receptores para C3b en los neutrófilos, lo que es un importante factor promotor de la quimiotaxis y fagocitosis.13
Los pilares del tratamiento son el control de los síntomas; el tratamiento de las infecciones y el control de las complicaciones.
Para mantener la integridad de la piel se recomiendan baños frecuentes con agua clorada, la aplicación de cremas emolientes y el uso de glucocorticoides sobre zonas inflamadas. En los casos necesarios la prescripción de antihistamínicos del tipo de antagonistas H1 es importante para el control del prurito.
Las infecciones bacterianas o fúngicas diseminadas se tratan de forma agresiva, y siempre que sea posible, con estudios dirigidos a identificar el agente causal y su sensibilidad a los diferentes antimicrobianos. Dentro de los antibióticos más utilizados se reporta el sulfametoxazol-trimetroprim (SMX-TMP), valioso en la prevención de las infecciones cutáneas estafilocócicas, sinusitis, otitis medias, infecciones respiratorias y neumonía, y el fluconazol es el agente de elección en las candidiasis mucocutánea. Dentro de las complicaciones pulmonares más frecuentes se encuentran los neumatoceles, aquellos que sean de gran tamaño deben de ser considerados como candidatos a cirugía.
Es importante controlar los niveles de calcio y vitamina D y usar suplementos orales ya que muchos pacientes presentan osteopenia y fracturas frente a traumatismos mínimos.
Se han comunicado casos aislados de mejoría en pacientes tratados con cimetidina (antagonistas de los receptores H2) ya que estos son importantes en la modulación de la quimiotaxis de los neutrófilos.
En casos seleccionados se ha utilizado con muy buenos resultados el interferón gamma recombinante humano. Algunos anticuerpos monoclonales, como el rituximab y omalizumab, se han empleado, aunque no se recomienda ninguno como tratamiento de primera línea. La utilización del trasplante de médula ósea sigue siendo controvertida, evidentemente la terapia génica es la solución en estos casos.
El uso de inmunosupresores como la ciclosporina A no está actualmente recogido en las guías clínicas; sin embargo, existen comunicaciones de casos con mejoría de quimiotaxis de los neutrófilos y descenso de las cifras de IgE sérica. Puede ser útil en pacientes con afectación muy grave que no responden a otras medidas.
La infusión con inmunoglobulinas, de forma intravenosa o subcutánea, no ha demostrado ser efectiva. Existen comunicaciones esporádicas de mejoría de algunos pacientes, especialmente en aquellos que no alcanzaban niveles de anticuerpos tras vacunaciones específicas. La administración de vacunas bacterianas atenuadas (BCG, tifoide oral) está contraindicada.2,14
En conclusión, SHIE-AD es una inmunodeficiencia primaria poco frecuente caracterizada por altos niveles de IgE, eosinofilia, abscesos cutáneos, eccema, candidiasis mucocutánea crónica e infecciones pulmonares recidivantes que contribuyen al desarrollo de neumatoceles y bronquiectasias; también asociada a alteraciones del tejido conectivo, esquelético, cerebral y vascular. Todas estas alteraciones se explican por la mutación asociada a STAT3 y a la alteración de la vía Th17. Se requiere de alto grado de sospecha clínica, siendo importante el manejo precoz de las infecciones que en general presentan escasa respuesta sistémica. Las alternativas terapéuticas están dirigidas a la prevención de la sepsis y el control de los síntomas.
REFERENCIAS BIBLIOGRÁFICAS
1. Leiva LE, Bezrodnik L, Oleastro M, Condino-Neto A, Costa-Carvalho BT, Sevciovic Grumach A, et al. Primary immunodeficiency diseases in Latin America: Proceedings of the Second Latin American Society for Immunodeficiencies (LASID) Advisory Board. Allergol Immunopathol (Madr). 2011; 39(2):106-110.
2. Guisado V, Fraile Rodríguez G, Arechaga Uriarte S. Síndrome de hipergammaglobulinemia IgE con infecciones recurrentes: patogénesis, diagnóstico y aproximación terapéutica. Rev Clin Esp. 2011; 211 (10):520-6 - DOI: 10.1016/j.rce.2011.04.009.
3. Estrada Reyes E, Hernández Román MP, Gamboa Marrufo JD, Valencia Herrera A, Nava Ocampo AA. Hypereosinophilia, Hyper-IgE Syndrome, and Atopic Dermatitis in a Toddler With Food Hypersensitivity. J Investig Allergol Clin Immunol. 2008;18(2):131-5.
4. Gamberale A, Moreira I, Bartoletti B, Cruz V, Bezrodnik L, Alberti F, et. al. Síndrome de Job asociado a tuberculosis miliar. MEDICINA 2014;74(4):311-4.
5. Tagle MT, Melys A, Castillo A, Norambuena X, Quezada A. Síndrome Hiper IgE: a propósito de tres casos clínicos. Rev Chil Pediatr. 2014;85(3):328-36.
6. Cantisanoa C, Díaza H, Balbaryskia J, Oleastro M, Quiroza H, Gaddia E. Inmunodeficiencia combinada con compromiso cutáneo asociada a mutación en DOCK8. Arch Argent Pediatr. 2014;112(4):e147-51. doi: 10.1590/S0325-00752014000400014.
7. Vásquez C, Martín Mateos MA, Giner MT, Sierra JI, Plaza AM, Díaz P, et. al. Otomastoiditis candidiásica y síndrome de hiper IgE. Allergol Immunopathol (Madr). 2004 Mar-Apr;32(2):82-5.
8. Montoya CJ, García de Olarte D. Síndrome de Hiperinmunoglobulinemia E con infecciones recurrentes (SHIEIR). CONSENSO LAGID, Grupo Latinoamericano de Inmunodeficiencias Primarias. 2001.
9. Navarrete Isidoro O, Flores Segovia J, Alonso Peces E, Alonso Viteri S, Losada Molina C, Ruiz Peña A, et. al. Absceso pulmonar con eosinofilia y aumento de IgE. Rev Patol Respir. 2009;12(supl 1):75-7.
10. de la Guardia Peña OM, Dr. Ustáriz García CR, García García MA, Morera Barrios LM. Síndrome de hiper IgE. Presentación de un caso. Rev Cubana Hematol Inmunol Hemoter. 2012;28(3):299-305.
11. Acosta PL. Un rol para Th17 en las enfermedades respiratorias. The Biologist (Lima). 2014;12(1):153-64.
12. D’Alessandro V, Pérez N. Diagnóstico temprano del síndrome de hiper IgE: un desafío. Arch Argent Pediatr. 2004;102(4):290-5.
13. Vega Orozco C, Hernández Velásquez L, Segura Méndez NH, Torres Salazar BA. Síndrome de hiper IgE. Diagnóstico y manejo oportunos. Revista Alergia México. 2008;55(1):38-45.
14. Campins M, Palacín PS. Vacunas recomendadas en pacientes afectados de una inmunodeficiencia primaria y sus familiares. An Pediatr (Barc). 2011;75(6):413.e1-22.
Recibido: marzo 02, 2015.
Aceptado: agosto 27, 2015.
Daily Pino Blanco. Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, CUBA. Tel (537) 643 8695, 8268
Email: rchematologia@infomed.sld.cu


{kind=link}
{kind=link}