










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La introducción de tecnologías como la secuenciación de nueva generación (NGS, por sus siglas en inglés) de ácido desoxirribonucleico (ADN) ha contribuido al descubrimiento de nuevos biomarcadores útiles en la definición del diagnóstico, pronóstico, predicción de la respuesta a terapias particulares y monitoreo de la enfermedad mínima residual. Todo ello depende de la detección o medición de uno o más biomarcadores específicos de diferentes enfermedades que representan alteraciones en vías genéticas o epigenéticas que controlan la proliferación, diferenciación o muerte celular.1
Dentro de las enfermedades hematológicas las neoplasias mieloproliferativas (NMP) representan un claro ejemplo del empleo de los biomarcadores en la práctica médica y de su importancia en el diagnóstico y tratamiento de los enfermos.
Las NMP constituyen un grupo fenotípicamente diverso de hemopatías malignas de origen clonal caracterizadas por una sobreproducción simple o multilineal de los elementos eritroides, mieloides y megacariocíticos; así como de una marcada predisposición a la trombosis, sangramiento y transformación leucémica. Aquí se incluyen tres NMP con características clínicas distintivas: la policitemia vera (PV), la trombocitemia esencial (TE) y la mielofibrosis primaria (MFP), conocidas como NMP clásicas BCR-ABL1 (o cromosoma Philadelfia) negativas.2
La PV se caracteriza por eritrocitosis frecuentemente combinada con trombocitosis o leucocitosis con supresión endógena de la producción de eritropoyetina y panmielosis en médula ósea. La TE presenta trombocitosis y médula ósea normocelular con proliferación megacariocítica. En la MFP prefibrótica se observan alteraciones en sangre periférica con proliferación granulocítica y megacariocítica en médula ósea, en ausencia de fibrosis reticulínica. En la MFP, además, se encuentra esta proliferación con signos de atipia acompañada por fibrosis de reticulina o colágeno de grado 2/3. También puede encontrarse desplazamiento anormal de stem cell con metaplasia mieloide (hematopoyesis extramedular en hígado o bazo).3
Las mutaciones somáticas en genes como JAK2 (Janus quinasa 2), MPL (Myeloproliferative Leukemia Protein) y CARL (calreticulina) se comportan como mutaciones drivers iniciadoras, responsables del fenotipo mieloproliferativo de las NMP. Estas son clasificadas así porque constituyen alteraciones genómicas que provocan una ventaja selectiva en una célula con capacidad para autorregenerarse, lo que conlleva a la formación de un clon de células mutadas.4,5 No obstante, se han encontrado mutaciones epigenéticas a nivel del gen TET2 (TET oncogene family member 2)4 y el haplotipo 46/16 que pueden preceder al fenotipo mieloproliferativo sin manifestaciones hematológicas. Existen estudios que han identificado mutaciones subclonales en los genes: TP53 (tumor protein p53), ASXL1 (Additional Sex Combs-Like 1), SRSF2 (serine/arginine-rich splicing factor 2), EZH2 (enhancer of zeste homolog 2), IDH1 e IDH2 (isocitrate dehydrogenase 1 and 2) y el propio gen TET2, que se asocian a la progresión de la enfermedad con alto riesgo de transformación leucémica y muerte prematura. 5,7-9 En total, se han identificado más de 20 mutaciones en las NMP.10
La Organización Mundial de la Salud (OMS), reconociendo el impacto de estas alteraciones moleculares en las NMP las incluyó dentro de los criterios diagnósticos mayores de estas enfermedades en sus dos últimas actualizaciones, (11,12) lo que confirma la robustez diagnóstica de estos biomarcadores.
Jak2
En el año 2005, cuatro grupos independientes describieron la presencia de la mutación V617F del gen JAK2 en las NMP.13-16 La proteína JAK2 derivada del gen con igual nombre, pertenece a la familia de tirosinas quinasa no receptoras JAK formada por 4 miembros, JAK1-3 y TYK2, que median la señalización de aproximadamente 60 citoquinas y hormonas.5 Su función biológica está determinada por su interacción con receptores específicos de citoquinas. Los reordenamientos del receptor inducidos por el ligando conducen a la fosforilación trans del dominio tirosina quinasa del JAK2, que resulta en estimulación de su actividad quinasa y progresión de la señal de transducción a través de las vías STATs (transductores de señal y activadores de la de transcripción), Ras-MAPK y PI3K/AKT, las cuales regulan la proliferación, apoptosis y diferenciación en células mieloides. Esta actividad se encuentra estrictamente regulada en varios niveles.5,17-18
La mutación JAK2V617F consiste en el cambio de una guanina por una timina en el nucleótido 1 849, localizado en el exón 14 del gen JAK2 en el cromosoma 9. Esta alteración genera el cambio de una valina (V) por fenila lanina (F) en la posición 617 de la cadena aminoacídica.19 Como consecuencia, se produce una ac tivación constitutiva de la proteína JAK2 en ausencia de la unión del ligando al receptor hematopoyético, que provoca una activación permanente de la vía de JAK/STATs. Esta mutación es la principal alteración molecular descrita en la NPM clásicas BCR-ABL1 negativas con una frecuencia del 95 % en la PV y del 55 al 60 % en la TE y MFP.19-22
Aunque esta mutación no es específica de ninguna de las NMP, su presencia proporciona evidencia de proliferación clonal y excluye cualquier consideración de causa reactiva. 23 Esta puede encontrarse en menos del 5 % de pacientes con leucemia mieloide aguda (LMA), síndromes mielodisplásticos, leucemia mielomonocítica crónica y otras hemopatías malignas. En la anemia refractaria con sideroblastos anillados y marcada trombocitosis puede hallarse con una frecuencia del 50 %.10 Aunque poco común, se han reportado casos de pacientes con esta mutación secundaria a la presencia del gen de fusión BCR-ABL1 en la leucemia mieloide crónica.24
En la MFP esta mutación se asocia con escasa supervivencia y edad avanzada, pero no hay un incremento significativo de riesgo de trombosis. También se relaciona con mayor conteo de leucocitos, nivel de hemoglobina y conteo de plaquetas. En la TE los pacientes presentan aumento de la incidencia de trombosis venosa y edad avanzada al debut, mayores niveles de hemoglobina, así como de leucocitos y menor conteo de plaquetas. En la PV la presencia de la mutación se vincula con altos niveles de hemoglobina y de neutrófilos, pero no hay correlación significativa con eventos trombóticos.25
Diversos estudios han estimado la carga alélica en la mutación V617F del JAK2 mediante la cuantificación por reacción en cadena de la polimerasa (PCR por sus siglas en inglés) en tiempo real, lo que constituye un marcador diagnóstico excelente para definir el pronóstico, complicaciones y evolución de las NMP.26,27) De acuerdo a esos estudios, los pacientes con TE presentan menor carga alélica que aquellos con PV, los pacientes con MFP presentan un rango intermedio entre estos dos y los aquejados de MF post-PV portan una mayor carga alélica que el resto.27
La baja carga alélica de la mutación JAK2V617F en la MFP se ha asociado con peor pronóstico, mientras que una carga alélica mayor del 50 % en la PV se vincula con un incremento del riesgo de transformación fibrótica y prurito.10 La estimación de la carga alélica permite evaluar la respuesta al tratamiento en estos enfermos.27
En el año 2007, se identificaron mutaciones en el exón 12 del gen JAK2 en solo 3-5 % de los casos de PV y eritrocitosis idiopática, negativas para JAK2V617F.28 Se han verificado clínicamente 27 alteraciones dentro de las que se incluyen sustituciones, deleciones y duplicaciones. Otras mutaciones del exón 12 han sido reportadas, pero se desconoce su significado clínico. La mutación más frecuente involucra la deleción de un marco de 6 nucleótidos del codón 542 al 543 y se encuentra presente en aproximadamente el 40 % de la PV negativas al JAK2V617F.19
Esas mutaciones inducen proliferación (independiente de citoquinas), de la expresión del receptor de eritropoyetina (EpoR) con incremento en los niveles de JAK2 fosforilado y STAT5.25
Los pacientes que portan mutaciones del exón 12 del gen JAK2 son significativamente más jóvenes, comparados con los portadores de la mutación JAK2V617F. Sus niveles de hemoglobina y hematocrito son mayores y la mayoría presenta conteo de leucocitos y plaquetas normales.25-29 Estos pacientes también pueden experimentar complicaciones trombóticas y transformación de la enfermedad a mielofibrosis o LMA de modo similar a los portadores de JAK2V617F.29
Otras mutaciones en los exones 3, 15, 14 y 16 (diferente de V617F) del gen JAK2 son excepcionalmente raras en las NMP y se han encontrado en diferentes enfermedades.5,19
Mpl
En el 2006, se describieron mutaciones que afectan al gen que codifica para el recep tor de la trombopoyetina, (TPO) MPL.30 La unión de la TPO al c-MPL (receptor de TPO) conduce a la activación del JAK2, el cual fosforila al c-MPL e inicia una cascada de eventos de señalización downstream que regulan la supervivencia, proliferación y diferenciación celular.19,25,31 Las mutaciones que derivan en la sustitución del aminoácido triptófano (W) producen un daño en la región autoinhibitoria del receptor y estimulación del receptor independiente del ligando,19,25,32 lo cual permite la activación de tirosinas quinasa y de factores de transcripción como STAT3 y STAT5, que a su vez provoca la transformación de células hematopoyéticas en clones independientes de citoquinas, lo que resulta en hiperplasia megacariocítica y fibrosis medular.19
Estas mutaciones se producen en el exón 10 del gen situado en el brazo corto del cromosoma 1(1p34) y afectan principalmente al aminoácido 515 y, con menor frecuencia, al 505. Se han definido varias alteraciones moleculares en esta región: W515L (más frecuente), W515K, W515A, W515R, S505N. También se han descrito en otros exones: V501A, Y252H y S204P. 19,25,33 La Y252H se encuentra en el dominio extracelular del MPL en pacientes con TE. 19 En general, las mutaciones en MPL son infrecuentes, aproximadamente entre el 3-15 % de los pacientes afectados de TE o MFP.19,21,25,33
Los pacientes en los que se identificaron estas variaciones tienen mayor riesgo de presentar complicaciones trombóticas y ser más dependientes de transfusiones que los pacientes positivos a la mutación JAK2V617F. Las alteraciones del MPL se relacionan con edad avanzada, alto conteo de plaquetas y elevados niveles de eritropoyetina (EPO); así como baja celularidad en médula ósea y cifras de hemoglobina disminuidas.25
Carl
En el 2013, se publicaron dos artículos que, aunque utilizaron enfoques diferentes para identificar afectaciones en el gen que codifica la proteína calreticulina, proporcionaron una fuerte evidencia genética de que las mutaciones en el gen CARL es importante en la patogénesis de la TE y MFP. 34,35
El gen CARL está localizado en el cromosoma 19 (19p13.2) y presenta 9 exones.10,25,36-38 La proteína CARL es una proteína multifuncional unida al Ca2+ con actividad de chaperona localizada fundamentalmente en el retículo endoplásmico (RE). Las mutaciones detectadas consisten en deleciones e inserciones que afectan al último exón del gen (el exón 9) y generan un cambio de estructura a un único marco de lectura alternativo que resulta en una nueva secuencia aminoacídica del dominio C-terminal. Además, en la proteína mutada está ausente la señal KDEL, lo que conduce a una dislocación parcial del CARL en el RE.4,25,36,38-40 En su mecanismo de acción el CARL activa al JAK2 en asociación con el c-MPLe induce trombocitosis en modelos en ratones. (36-40)
Las mutaciones en CARL representann la segunda alteración molecular con mayor prevalencia después de la mutación JAK2V617F.4,25 Su frecuencia se estima en 49 % en la TE y el 74 % en la MFP sin JAK2/MPL mutado.41Aunque se han detectado más de 50 tipos diferentes de mutaciones en el exón 9 del CARL, más del 80 % corresponden a las mutaciones tipo 1 (deleción de 52 pb) y tipo 2 (inserción de 5 pb).4,25,41) Las mutaciones CARL no tipo 1/2 se clasifican en variantes similares al tipo 1 o 2 (“type like-1” o “type like-2”) sobre la base de su semejanza estructural con cada una de ellas.25 Las mutaciones tipo 1 son las más comúnmente encontradas tanto en la MFP como en la TE y se asocian con mayor supervivencia de los pacientes en comparación con las mutaciones tipo 2 y la mutación JAK2V617F.10,25,41
En diversas investigaciones se ha observado que los pacientes con TE positivos a mutaciones del CARL, comparados con los positivos al JAK2V617F, presentan menores niveles de hemoglobina y de leucocitos; mayor conteo de plaquetas y menor riesgo de trombosis. 10,25,41-42 Sin embargo, otros estudios señalan un riesgo relativamente alto de transformación mielofibrótica (especialmente asociado con la mutación tipo 1).3) En la MFP las mutaciones CARL se relacionan con pacientes más jóvenes, mayor conteo de plaquetas y menor incidencia de anemia y leucocitosis con relación a las mutaciones de JAK2 y MPL.10,25,41 Los pacientes son menos dependientes de transfusiones sanguíneas y tienen mayor supervivencia que los portadores de otras mutaciones.25
NMP triple negativas
Alrededor del 10 % de los pacientes con TE y aproximadamente del 5 al 10 % de aquellos con MFP reúnen los criterios diagnósticos establecidos por la OMS para las NMP, pero no se identifican en ellos ninguna de las tres mutaciones drivers aquí analizadas. Este grupo de pacientes se definen como triples negativos.3,10,43 En algunos de ellos con TE, se han identificado mutaciones atípicas del gen MPL y variantes de mutaciones del gen JAK2.33,43-44 En general, la TE triple negativa se comporta de manera más benigna con baja incidencia de eventos vasculares. En cambio, la MFP triple negativa se muestra como una neoplasia mieloide agresiva con características mielodisplásicas prominentes y alto riesgo de progresión leucémica.3,43
Algoritmo diagnóstico
El algoritmo diagnóstico de los biomarcadores en las NMP tiene en cuenta dos aspectos fundamentales: la frecuencia de distribución del JAK, CARL y MPL en las NMP BCR-ABL1 negativas y el hecho de que estas se consideran mutuamente excluyentes entre sí (aunque existen reportes aislados de su coexistencia) (fig. 1). 20,45-46 La OMS recomienda, en ausencia de estos tres biomarcadores principales, la búsqueda de mutaciones acompañantes para determinar la naturaleza clonal de la enfermedad (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1).12
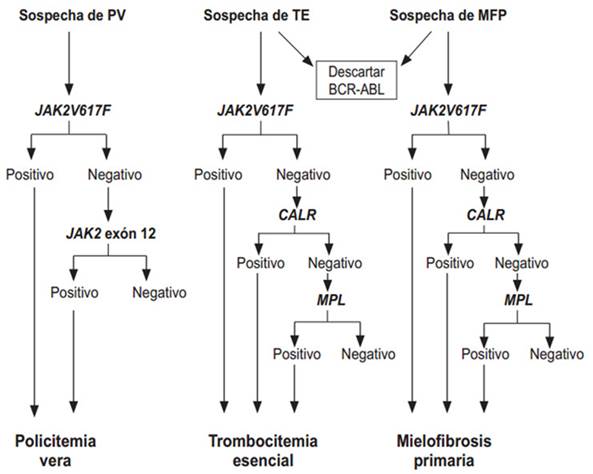 Leyenda: PV: Policitemia vera; TE: trombocitemia esencial; MFP: mielofibrosis primaria; BCR-ABL: cromosoma Filadelfia; JAK2: Janus quinasa 2; JAK2V617F; CALR: calreticulina; MPL: Myeloproliferative Leukemia Protein (Tomado y modificado de Sieza Y, et al. HEMATOLOGÍA. 2018; 22(2): 151-6) 20
Leyenda: PV: Policitemia vera; TE: trombocitemia esencial; MFP: mielofibrosis primaria; BCR-ABL: cromosoma Filadelfia; JAK2: Janus quinasa 2; JAK2V617F; CALR: calreticulina; MPL: Myeloproliferative Leukemia Protein (Tomado y modificado de Sieza Y, et al. HEMATOLOGÍA. 2018; 22(2): 151-6) 20
Fig. 1- Algoritmo diagnóstico de las NMP BCR-ABL1 negativas
Aunque el estudio molecular proporciona información valiosa para el diagnóstico y seguimiento de las NMP y muestra evidencia de clonalidad, no logra diferenciar entre cada una de ellas. (12,47 Por esto, se requiere de la adecuada aplicación del método clínico para llegar a un diagnóstico certero con ayuda de otros exámenes complementarios como la biopsia de médula ósea, especialmente en la TE y la MFP, para identificarlas entre sí. 12

