










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La macroglobulinemia de Waldenström (MW) se define como un linfoma linfoplasmocítico con compromiso de la médula ósea (MO) y gammapatía monoclonal IgM.1 La MW se caracteriza por presentar hiperviscosidad como principal manifestación clínica, así como linfadenopatías y hepatoesplenomegalia. En los Estados Unidos de Norteamérica se notificó una incidencia anual de 3,8 casos por millón de habitantes durante el periodo 1988-2007, con aumento en relación a la edad.2 La MW es más frecuente en hombres caucásicos, la edad media de presentación es entre los 60-70 años; y representa la segunda entidad más frecuentemente relacionada a la amiloidosis de cadenas ligeras (AL).3
La MW tiene un curso indolente y una mediana de supervivencia de 5 años. La mutación somática MYD88 L265P está presente en más del 90 % de los casos y forma parte actualmente de los estudios de diagnóstico molecular.4,5 Alrededor del 70 % de los casos de MW presentan síntomas, los más frecuentes son los relacionados a fatiga, sangrado, síntomas B y linfadenopatías periféricas.6 No obstante, pueden presentarse síntomas relacionados al depósito de la paraproteína M, como: síndrome de hiperviscosidad, amiloidosis sistémica, crioglobulinemia y neuropatía periférica.5
Resulta importante diferenciar esta enfermedad del mieloma múltiple, pues ambas pueden afectar la médula ósea; sin embargo, la MW no presenta lesiones óseas ni hipercalcemia. Para el diagnóstico, se debe demostrar una infiltración de células linfoplasmocíticas mayor al 10 % en la MO con fenotipo IgM + de superficie, CD19+, CD20+, CD22+, CD5+ raramente, CD10-, CD23-, y aumento de mastocitos. La IgM se encuentra aumentada principalmente en el suero mas no en la orina debido a la mínima excreción renal de las inmunoglobulinas.4
La AL representa una manifestación infrecuente de la MW y está definida como el depósito extracelular de fibrillas amiloides formadas por subunidades proteicas de bajo peso molecular. Fisiopatológicamente es originada por el mal plegamiento de las cadenas ligeras, y ulteriormente de las inmunoglobulinas, las cuales se acumulan y depositan en la matriz extracelular de varios órganos.7 Cuando el depósito es a nivel miocárdico, resulta ser un factor crucial para el pronóstico de la enfermedad.
El objetivo de esta presentación es describir un caso de diagnóstico desafiante sobre MW con amiloidosis primaria cardiaca.
Caso clínico
Paciente masculino de 77 años, con antecedentes de diabetes mellitus controlada con hipoglucemiantes orales e hipotiroidismo en tratamiento, y con antecedente familiar de cáncer esporádico. Fue admitido por astenia y disfonía de 3 años de evolución. Previo a su ingreso se agregó edema bilateral de miembros inferiores, ortopnea y disnea. Al ingreso, presentó efusión pleural bilateral y palidez mucocutánea; el resto del examen resultó anodino.
Entre los exámenes auxiliares se encontró anemia macrocítica hipercrómica, anisocitosis, neutropenia leve, plaquetopenia leve. Mediante estudios de imágenes se confirmó derrame pleural, el cual fue drenado con mejoría transitoria. Una tomografía de tórax posterior sin contraste evidenció neumotórax laminar en hemitórax derecho, así como pequeñas áreas focales de proceso parénquimal de aspecto inflamatorio a nivel bibasal. Dentro de los exámenes en cardiología se destacó la presentación de un compromiso de patrón granular a nivel miocárdico y efusión pleural de etiología no filiada con realización de pleurodesis sin mejoría. El valor basal de la porción N-terminal del pro-péptido natriurético tipo B (NT-pro-BNP) fue de 812 pg/mL, el cual aumentó progresivamente hasta un valor máximo de 2905 pg/mL antes del inicio del tratamiento (Fig 1 y 2).
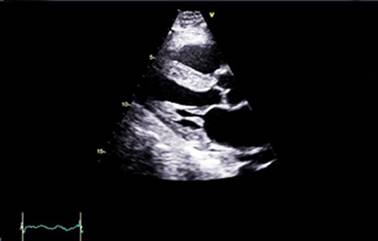
Fig. 1 - Ecocardiograma transtorácico con vista paraesternal de eje largo. Miocardio con patrón ecogénico granular e hipertrofia concéntrica moderada del ventrículo izquierdo a predominio del septum basal.
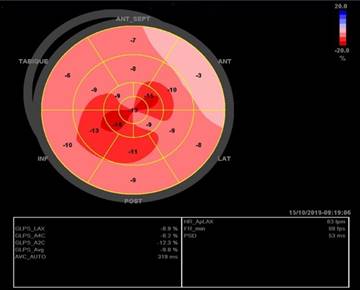
Fig. 2 - Strain longitudinal global. Patrón ecocardiográfico típico de strain longitudinal global anormal en amiloidosis cardíaca con reducción del strain pico sistólico basal y preservada a nivel apical que se muestra como un patrón característico en el mapa de ojo de buey. Este patrón puede ser una clave ecocardiográfica cuando otros signos son sutiles o están ausentes.
En la ecocardiografía transtorácica se evidenció disfunción diastólica de ventrículo izquierdo de tipo II con presiones de llenado elevadas, insuficiencia mitral leve e insuficiencia tricúspidea moderada.
En los exámenes de laboratorio se obsevó disproteinemia leve (4,4 g/dL) con proteinograma electroforético sérico en lo que se apreció un pico monoclonal en región gamma: 2,15 g/dL; β2-microglobulina: 2,5 mg/L, inmunofijación sérica y en la orina con banda kappa libre y cuantificación de cadenas ligeras elevada para kappa libre: 328,15 mg/L, lambda libre disminuida: 3,82 mg/L e índice kappa/lambda 85,9 mg/L y dosaje de IgM elevado 25,4 g/L (Fig. 3).
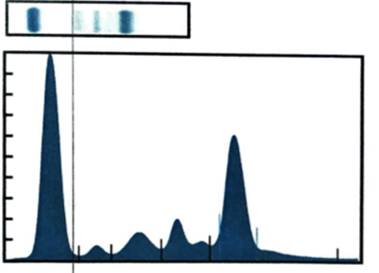
Fig. 3 - Trazado electroforético. Se observa banda de pico monoclonal en la región de las gammaglobulinas.
La evaluación del líquido pleural resultó ser un trasudado y fue negativo para Mycobacterium, así como para otros gérmenes. El estudio de la biopsia de la pleura demostró pleuritis crónica inespecífica con presencia de células mesoteliales, la tinción de rojo congo resultó positiva para depósito de sustancia amiloide (Fig. 4). Además, se empleó la misma tinción en la piel y en la grasa periumbilical, con resultados negativos.
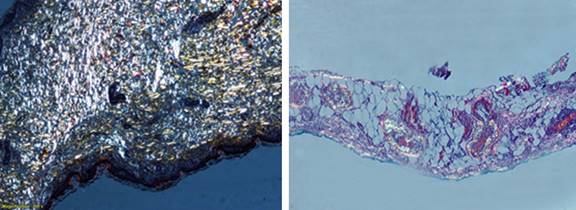
Tanto la citomorfología como la citometría de flujo en la médula ósea confirmaron la sospecha de MW, con 12 % de infiltración por células linfoplasmocíticas.
Por los hallazgos descritos anteriormente se diagnosticó al paciente con amiloidosis cardiaca por MW. Posteriormente, se inició tratamiento con bortezomib, dexametasona y rituximab con mejoría clínica y resolución de los síntomas.
Discusión
El cuadro clínico comenzó con una disfonía por parálisis de las cuerdas vocales; sin embargo, el 50 % de los pacientes con MW inician el cuadro con síntomas constitucionales. La literatura consultada señala que la IgM puede tener especificidad por una glucoproteína asociada a mielina (MAG), lo que conlleva a que los pacientes con MW desarrollen una neuropatía periférica característica. Se ha planteado que este síntoma puede preceder a la neoplasia, lo que concuerda con la aparición atípica de nuestro caso. La teoría propuesta por los expertos sería que una infección viral puede provocar una respuesta de anticuerpos que reaccionen de forma cruzada con un componente de tejido normal. Asimismo, se suele presentar miocardiopatía restrictiva, síndrome nefrótico, hepatomegalia y macroglosia.4,8
Está descrito que el tiempo invertido en consultas médicas, desde el primer contacto en servicios de salud al diagnóstico, es de aproximadamente 2 años e incluye de 6 a 7 evaluaciones previas en promedio.8,9 Nuestro paciente acudió a más de 5 especialistas de diferentes instituciones durante 3 años sin llegar a un diagnóstico claro. Este caso representa un reto clínico y terapéutico por la presentación variada de signos y síntomas según el órgano afectado.
La identificación de la enfermedad se hace mediante tinción con rojo congo o azul alcian sulfatado, inmunohistoquímica o imágenes cardiacas positivas con amiloidosis extracardiaca conocida.10 Sin embargo, una vez realizadas las pruebas existe el riesgo de encontrarse con falsos negativos si solo se realiza la evaluación en la grasa periumbilical, pues su sensibilidad llega al 75 %. Por lo general, el 59 % de los casos tienen depósitos de amiloide en la MO y en la grasa subcutánea, mientras que un 13 % presenta resultados positivos solo en la MO, cuya sensibilidad alcanza el 69 %.11 Actualmente, se recomienda hacer un análisis exhaustivo ante la sospecha del diagnóstico, sobre todo si se manifiesta con disfunción diastólica por infiltración de las fibrillas proteicas, lo que restringe el movimiento de las paredes ventriculares y desencadena una insuficiencia cardiaca derecha. Asimismo, pueden aparecer arritmias, coronariopatías, valvulopatías, entre otras afecciones cardiacas.12
En este caso se encontró compromiso cardíaco, cuyo grado de afectación determina el pronóstico del paciente, que generalmente es desfavorable.13 Para su evaluación, se emplean tres biomarcadores: el pro-BNP, la troponina sérica y la diferencia entre las cadenas ligeras libres de inmunoglobulinas involucradas y no involucradas; los cuales funcionan como predictores de respuesta. Con estas 3 pruebas positivas, clasificamos a nuestro paciente en estadio 4 con una supervivencia global de 6 meses.8
Al cuadro clínico del paciente se agregó: astenia, disnea progresiva, ortopnea y edema bilateral de miembros inferiores con fóvea en región pretibial característicos de afección cardiaca que suele presentarse como manifestación del síndrome de hiperviscosidad; así como cuadros de efusión pleural.9 No obstante, al examen físico no presentó hallazgos sugerentes de amiloidosis como púrpura periorbitaria (presente en un 15 % de pacientes), macroglosia (en 1 de cada 8 personas), hepatomegalia (20 % de pacientes) y síncope. En los pacientes con MW, los exámenes de rutina comúnmente muestran anemia normocítica normocrómica, presencia de fenómeno de rouleaux y prueba de Coombs positivo; sin embargo, nuestro paciente presentó anemia macrocítica hipercrómica, sin hallazgos de alteraciones morfológicas en el extendido de sangre periférica.4,14 En estudios de casos de MW, se suele encontrar dilatación de venas retinianas y segmentación vascular con múltiples hemorragias retinales como consecuencia de la hiperviscosidad; además, suelen haber alteraciones óseas. Sin embargo, en nuestro caso, no se presentaron las alteraciones anteriormente descritas, lo que dificultó aún más la sospecha diagnóstica.4,15
A pesar de la aparición de nuevas herramientas, el diagnóstico tardío es una desventaja importante para la supervivencia; cerca del 40 % de pacientes no logra sobrevivir al primer año posterior al diagnóstico por el grado de afectación miocárdica.10 Es por esa razón, que su manejo es paliativo de las manifestaciones cardiacas. En este caso se brindó un diurético (espironolactona). El tratamiento base considerado en amiloidosis AL es el de preservar y mejorar la función del órgano afectado porque la mayoría de los pacientes no son candidatos al trasplante del órgano perjudicado y deben ser tratados con quimioterapia sistémica tradicional, la cual consiste en un régimen de ciclofosfamida, bortezomib y dexametasona. Este régimen de tratamiento es indispensable ya que tiene una alta tasa de respuesta y logra revertir la enfermedad hasta en 17 % de los pacientes.16 Si se considera el estadío de enfermedad, el paciente tiene una probabilidad de supervivencia de 14 % a los 5 años, y ajustándose a las manifestaciones clínicas de este, se inició tratamiento con bortezomib, dexametasona y rituximab.17 La literatura internacional recomienda ante una potencial recaída, el uso de Ibrutinib en MW con MYD88 mutado y CXCR4 salvaje como segunda línea de terapia por su alta tasa de respuesta.4,18 Recientemente, se ha demostrado que el daratumumab, un anticuerpo monoclonal anti CD38, retrasa el crecimiento tumoral in vivo en ratones con MW, gracias a su alto efecto citotóxico sobre células MW con o sin resistencia a ibrutinib.19)
En esta enfermedad el diagnóstico temprano es una ventaja importante para la supervivencia. Es por esa razón, que su manejo es paliativo de las manifestaciones cardiacas. El presente caso pone en manifiesto un reto diagnóstico, en el cual se deben tomar en cuenta las etiologías menos frecuentes de insuficiencia cardiaca.

