










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Behcet (SB), o enfermedad de Behcet (EB) es un proceso autoinflamatorio crónico, que cursa con periodos de remisión, de baja frecuencia y etiología desconocida.1,2,3 Es una vasculitis neutrofílica que afecta arterias y venas de todos los calibres, con alteración de la función endotelial, que se expresa clínicamente con lesiones orgánicas variadas, en las que predominan las lesiones mucocutáneas y oculares.2,3) Comparte características comunes con enfermedades autoinmunes, autoinflamatorias y espondiloartropatías (MHC - opatías).3)
Su diagnóstico es clínico, y se realiza de acuerdo a criterios del grupo internacional de estudio (ISG, del inglés International Study Group) de la enfermedad de Behcet.1
Este grupo definió como criterios clínicos para el diagnóstico la presencia de úlceras orales recurrentes (aftosas o herpetiformes), sin otra causa demostrada, al menos 3 veces en un periodo de 12 meses; asociadas a 2 de los siguientes signos:
Úlceras genitales recurrentes aftosas o cicatrizales.
Lesiones oculares: Uveítis anterior o posterior, células en el vítreo, vasculitis retinal, observadas por un oftalmólogo.
Lesiones en piel: Eritema nodoso, pseudofoliculitis, lesiones papulopustulares o nódulos acneiformes (en pacientes adultos no tratados con esteroides).
"Test de patergia" positivo: Leído por un médico dentro de las 24-48 horas de realizado.
Posteriormente un grupo internacional integrado por representantes de 27 naciones, propuso otorgar 2 puntos a las lesiones oculares y a las lesiones aftosas orales y genitales y 1 punto a las lesiones de piel, del sistema nervioso central, las manifestaciones vasculares y al test de patergia positivo. Para realizar el diagnóstico de EB el enfermo debe tener una puntuación ≥ 4 puntos.4
Las afectaciones más graves suelen ser las oculares y las vasculares. Las afectaciones menos frecuentes son las de grandes vasos gastrointestinales, pulmonares y renales.2
La frecuencia del síndrome de Behcet es similar en ambos sexos, aunque existen variaciones en diferentes regiones geográficas. En países como Estados Unidos de América, Japón y Corea predomina en el sexo femenino y en la región del mediterráneo oriental en el masculino. La severidad suele ser mayor en el sexo masculino.1,2
En su etiología intervienen factores genéticos, infecciosos (virales y bacterianos) e inmunológicos.1
Entre los más importantes se señalan la asociación con el fenotipo HLA (del inglés Human Leucocyte Antigens) de los pacientes, específicamente con la molécula HLA-B*51 y la activación del endotelio vascular.2
En diversos estudios se han descrito alteraciones del sistema inmune en el SB, entre ellas: alteración en el número y activación de los linfocitos; inversión del índice CD4+/CD8+; expansión oligoclonal de células T; elevación de los niveles séricos de algunas interleucinas (IL) y receptores (R), tales como la IL-8 (citocina que produce activación de los neutrófilos) y de las citocinas proinflamatorias IL-1, IL-6, Il-12, Interferon (INF), Factor de necrosis tumoral (FNT), y sIL-2R. Estos hallazgos sugieren el predominio de un patrón de respuesta immune de tipo Th 1.1
El tratamiento de la enfermedad es sintomático, con el objetivo de reducir los síntomas, y prevenir las complicaciones. Se emplea tratamiento tópico y sistémico, que incluye el empleo de esteroides, inmunosupresores y agentes biológicos.1
Presentación del caso
Paciente masculino de 39 años de edad, con diagnóstico clínico de enfermedad de Behcet desde hace 20 años, dado por la presencia de signos mayores y menores de la enfermedad: aftas orales y genitales recurrentes, lesiones papulosas en piel de miembros superiores e inferiores, episodios recurrentes de vasculitis en miembros inferiores, uretritis y artralgia. No presenta signos de uveítis. El paciente refiere la presencia de síntomas desde los 7 años de edad.
En el momento del estudio se encontraba en condiciones basales de la enfermedad.
Antecedentes patológicos familiares: madre y 2 hermanos con aftas orales y genitales y artralgias recurrentes.
Estudios complementarios:
Eritrosedimentación: 23 mm/h
Inmunoglobulinas: IgG: 10,20 g/L (VR: 7,81-15,30); IgM: 2,16 g/L (VR: 0,69-2,69); IgA: 3,50 g/L (VR: 0,83-4,6).
Anticuerpos antinucleares (ANA): negativo; Anticuerpos anticitoplasma de neutrófilos (ANCA): negativo.
Componentes del Complemento C3: 1,32 (VR: 0,89-1,81) C4: 0,292 (VR: 0,165-0,380)
Inmunocomplejos circulantes (ICC):12 (VR < 16 µg eq/mL)
Proteina C reactiva: negativa; Factor reumatoideo (FR): negativo
Análisis de subpoblaciones linfocitarias en citómetro Gallios, Beckman coulter, Software Kaluza versión 1.2: CD3+ CD4+ 35,98 % (VR:28-57); CD3+ CD8+: 15,21 % (VR:13- 42); CD19+ se observa doble población de linfocitos B: 9,01 % , 2,98 % (VR:6-19) (Fig.1); CD56+: 24,13 % (VR:7-31 )
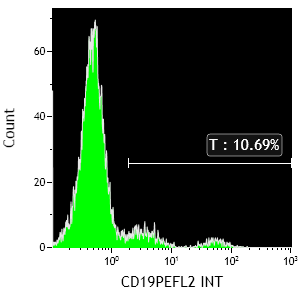
Marcadores de activación determinados en paralelo con la muestra de un individuo adulto sano, el cual se empleó como control normal (CN).
CD4+ CD25+: 0,76 % (CN: 13,18 %); CD19+ CD25+: 0,08 % (CN: 0,62 %); CD56+ CD25+: 0,02 % (CN: 0,65 %)
Se logró la remisión de los síntomas y signos con tratamiento esteroideo. Al inicio del diagnóstico se indicó tratamiento con Prednisona 40 mg/día y después de la remisión clínica se redujo progresivamente, a razón de 5 mg cada 3 días, hasta alcanzar una dosis de mantenimiento de 10 mg/día. Con dosis inferiores aparecen úlceras orales y genitales. Durante las crisis se administra tratamiento con prednisona 30 mg/día.
Discusión
La tasa de prevalencia de síndrome de Behcet, por número de habitantes, varía en las diferentes regiones geográficas: (20-421/100 000 en Turquía, 13-17/100 000 en Japón, Corea y China, 0,5-3/100 000 en Europa, 1/170 000 en Estados Unidos de América.1,2 En Cuba no existen estudios de prevalencia de esta entidad.2
El síndrome de Behcet puede presentarse a cualquier edad, pero es más frecuente en adultos jóvenes de entre 20 y 30 años de edad,1,2 aunque en un estudio realizado en Cuba en 2016, la edad promedio fue superior.5) En el caso de estudio, los síntomas y signos iniciales de SB aparecieron en la infancia, aunque el diagnóstico se realizó a los 19 años de edad.
El paciente presentó las manifestaciones clínicas más comunes de la enfermedad: úlceras aftosas orales y genitales, afecciones de la piel y artritis, que satisfacen los criterios diagnósticos del grupo internacional de estudio de la EB y del consenso internacional de 2013, en este último caso con un score de 4 puntos.1,4,5
La mayoría de los casos de SB son esporádicos,2 pero algunos estudios sustentan un posible origen genético del síndrome, entre estos: Gül y otros, hallaron que en 1 de cada 10 familias existe otro miembro con la enfermedad.6) Molinari y otros, encontraron evidencias de herencia autosómica recesiva en casos pediátricos7) y de Menthon, en metanálisis realizado, enfatizó la fuerte asociación del SB con el alelo HLA-B51.8
Algunos investigadores sugieren que, debido a su fuerte asociación a la molécula HLA-B*51, el SB pertenece al grupo de las llamadas enfermedades HLA clase I - patías que incluye entidades como la espondilitis anquilosante y la psoriasis asociadas al HLA-B*27 y HLA-C*0602, respectivamente y que la interacción de receptores en las células del sistema inmune con el péptido del sistema mayor de histocompatibilidad podría desencadenar las manifestaciones inflamatorias observadas.9
Los estudios más recientes sobre la etiopatogenia del SB sugieren que agentes infecciosos tales como el Streptococcus sanguinis o las diferencias en la composición de la microbiota salival o intestinal pueden provocar inflamación por mecanismos de la inmunidad innata, en individuos genéticamente susceptibles, la que subsecuentemente se mantiene y amplifica por la respuesta inmune adaptativa.3) Otro factor a considerar es la presencia de péptidos endógenos alterados, como consecuencia de polimorfismos de los genes que codifican la aminopeptidasa retículo endoplásmica 1 (ERAPI 1, del inglés endoplasmic reticulum aminopeptidase 1). De este modo, la presentación por la molécula HLA-B * 51 de agentes infecciosos y/o péptidos endógenos alterados juega un papel clave en la patogenia del SB, provocando una alteración del balance de las células T, disminución de la regulación por las T reguladoras y expansión de las Th1 y las Th17.3,9
Lucherinia y otros, plantean que las células Th 17 juegan un papel de pivote en la patogenia del SB y que los altos niveles de amiloide A sérico constituyen un marcador potencial de actividad de la enfermedad.10 A partir de su estudio sugieren que en el SB se produce más que una diferenciación de CD4+ naive a Th17, una polarización a Th17.
Sonmez y otros, encontraron en pacientes con enfermedad activa niveles altos de actividad Th17 y respuesta Th1 y bajos niveles de IL-35, en relación a controles y enfermos no activos, por lo que sugieren que puede existir plasticidad entre las células Th17 y las T reguladoras, de acuerdo al estado de actividad de la enfermedad.11
La mayoría de los autores plantea que las células asesinas naturales o NK (del inglés natural killer) pueden jugar un papel inmunoregulatorio en el SB a través de la interacción de su receptor KIRK3DL1 con su ligando Bw4, en la molécula HLA-B*51.12,13 Otras hipótesis alegan que alteraciones de la molécula HLA-B*516 o polimorfismos de los receptores KIR pueden provocar directamente la inflamación.14
También se reportan como factores predisponentes del SB las alteraciones en los genes codificadores de citocinas. Recientemente Zarraby y otros, detectaron niveles significativamente bajos de la interleucina anti inflamatoria Il-38, perteneciente a la familia de la Il-1, en individuos con afectación ocular y test de patergia positivo, en comparación con controles normales.15
Los antecedentes familiares del paciente: madre y dos hermanos afectados, sugieren posible predisposición genética, por lo que sería conveniente realizar un estudio familiar de los genes de histocompatibilidad y codificadores de citocinas.
Las subpoblaciones linfocitarias se encontraron dentro de los valores referenciales y no se observaron marcadores de activación linfocitaria. El enfermo objeto de estudio no se encontraba en fase activa de la enfermedad, lo que explica la ausencia de alteraciones en el estudio, sin embargo, la presencia de una doble población de células B, puede ser indicio de activación policlonal y la existencia de una población de linfocitos B de autoreconocimiento.
No existen alteraciones de laboratorio típicas del SB. Los reactantes de fase aguda, y el estudio de marcadores de autoinmunidad suelen ser negativos, lo que coincide con los hallazgos de este estudio. Sin embargo algunos autores han observado elevación de los valores de reactantes de fase aguda en pacientes con vasculitis de grandes vasos y otros describen niveles moderadamente elevados de eritrosedimentación y proteína C reactiva, sin correlación con la actividad de la enfermedad.2,5
Algunos estudios muestran elevación de la concentración de las inmunoglobulinas séricas y de los niveles de inmunocomplejos, pero con autoanticuerpos como el FR, ANA, anticardiolipina y ANCA negativos.2
Otros autores refieren la presencia de autoanticuerpos específicos contra proteínas de la mucosa, células endoteliales, LDL (del inglés, low density lipoprotein) oxidada y el antígeno tropomiosina en la uveítis.2
En el caso que se presenta, los niveles de inmunoglobulinas, componentes C3 y C4 del complemento e ICC se encontraron dentro de los valores referenciales y no se observó presencia de ANA, ANCA, ni FR, por lo que no se detectan evidencias humorales de autoinmunidad.
El tratamiento incluye medicamentos de uso tópico y sistémico, entre estos los más empleados son los esteroides, los inhibidores del factor de necrosis tumoral y los inmunosupresores.1,2) El tratamiento con prednisona fue eficaz en el caso de estudio, lográndose la remisión clínica con una dosis de mantenimiento de 10 mg/día.
El estudio apoya el criterio de que, en la EB en condiciones basales, no se detectan marcadores humorales de autoinmunidad, alteraciones de los valores de las subpoblaciones linfocitarias, ni evidencias de activación linfocitaria, pero no se puede excluir la presencia de una población de linfocitos B de autoreconocimiento.

