










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Las neoplasias mieloproliferativas clásicas BCR-ABL (NMP punto de ruptura en la región Abelson, por sus siglas en inglés) negativas son las enfermedades más frecuentes entre los trastornos mieloproliferativos crónicos.1) Se clasifican en policitemia vera (PV), trombocitemia esencial (TE) y mielofibrosis primaria (MF).2
La trombocitemia esencial se caracteriza por una trombocitosis mantenida en sangre periférica e hiperplasia de megacariocitos maduros en médula ósea,2,3) y clínicamente por una tendencia a complicaciones trombóticas, hemorrágicas o ambas, junto con marcadores moleculares y características histológicas específicas, con el consecuente riesgo a largo plazo de la transformación a una mielofibrosis o a una leucemia mieloide aguda (LMA). Tiene un pronóstico favorable, con una supervivencia de 19,8 años.2,3
La incidencia global de TE es de 0,6/100 000 pacientes/año (de 0,004 a 0,9),4) sin embargo, la incidencia anual estimada en los Estados Unidos de América es de 2,5 casos por 100 000 habitantes, mientras que la prevalencia es de 24 casos por 100 000 habitantes.4) Al diagnóstico se presenta con una mediana de edad entre los 55-60 años, tiene una incidencia más baja entre los hombres que entre las mujeres, en una proporción de 1,6 mujeres / 1 hombre.4
En Cuba, la incidencia de la TE y de otras NMP se desconoce. El anuario estadístico de salud agrupa a este conjunto de neoplasias en el apartado: otros tumores del tejido linfático y de los órganos hematopoyéticos,5) sin embargo, no se especifica la contribución de cada una de ellas por separado a la morbilidad y mortalidad global.
El diagnóstico de la TE se ha basado en criterios diagnósticos, los primeros propuestos por el Grupo de estudio de la PV (PVSG, por sus siglas en inglés) en 1986.6 La Organización Mundial de la Salud (OMS) estableció nuevos criterios en el 2001, y en el año 2008 fueron modificados,6 al combinar resultados de laboratorio con características morfológicas y hallazgos genéticos y moleculares. En la actualidad la TE se basa en los criterios modificados por la OMS en el 2016.7
Con el descubrimiento en el 2005 de la mutación del gen Janus quinasa 2 (JAK2V617F) se determinó una de las principales alteraciones moleculares en la TE. La mutación consiste en el cambio de una guanina por una timina en el nucleótido 1849 localizado en el exón 14 del gen JAK2 en el cromosoma 9 y se genera el cambio de una valina (V) por fenilalanina (F) en la posición 617 de la cadena aminoacídica, como consecuencia.8) Se produce una activación constitutiva de la proteína JAK2 en ausencia de la unión del ligando al receptor hematopoyético, que provoca una activación permanente de la vía de JAK/STATs; del 55 al 60 % los pacientes presentan esta mutación molecular.8
En el año 2006 se describe las mutaciones que afectan al gen que codifica para el receptor de la trombopoyetina (TPO) MPL (myeloproliferative leukemia protein por sus siglas en inglés).8) La unión de la TPO al c-MPL (receptor de TPO) conduce a la activación del JAK2, el cual fosforila al c-MPL e inicia una cascada de eventos de señalización que regulan la supervivencia, proliferación y diferenciación celular.8,9) La mayoría de las mutaciones del MPL se producen en el exón 10 del gen situado en el brazo corto del cromosoma 1 (1p34) y afectan por lo general al aminoácido 515 y con menor frecuencia al 505. Se presenta en el 5 al 10 % de los casos de TE.8,9
En el 2013 se publicaron dos artículos que, aunque utilizaron enfoques diferentes para identificar mutaciones en el gen que codifica la proteína calreticulina (CALR), proporcionaron una fuerte evidencia genética de que las mutaciones en el gen CALR tienen un papel importante en la patogénesis de la TE.10) El gen CALR está localizado en el cromosoma 19 (19p13.2) y presenta 9 exones. La CALR es una proteína multifuncional unida al calcio activado (Ca2+) con actividad de chaperona, localizada en el retículo endoplásmico (RE).9,10) En su mecanismo de acción el CALR activa al gen JAK2 en asociación con el c-MPL e induce trombocitosis en ratones. Su frecuencia se estima en un 49 % y representa la segunda alteración molecular con mayor prevalencia después de la mutación JAK2V617F en la TE.8,9,10
Alrededor del 10 % de los pacientes con TE reúnen los criterios diagnósticos establecidos por la OMS, pero no se identifican en ellos ninguna de las tres mutaciones conductoras aquí analizadas. Ese grupo de pacientes se definen como triples negativos.8,9,10
La combinación de los estudios clínicos, morfológicos y moleculares permite realizar una adecuada estratificación pronóstico, indicar un tratamiento y seguimiento personalizado e identificar factores que de manera individual contribuyan a la aparición de complicaciones que atenten contra la calidad de vida y supervivencia prolongada de estos pacientes.
El objetivo del estudio fue caracterizar a los pacientes adultos con trombocitemia esencial atendidos en el Instituto de Hematología e Inmunología (IHI).
Métodos
Se realizó un estudio observacional, descriptivo, longitudinal y retrospectivo en el que se incluyeron los pacientes adultos con diagnóstico de TE atendidos en el IHI desde enero del 2010 hasta enero del 2020. El universo de estudio estuvo conformado por 40 pacientes, mayores de 18 años de edad en el momento del diagnóstico de acuerdo a los criterios definidos por la OMS del 2008.6
Las variables demográficas estudiadas fueron la edad, sexo y color de la piel.
Las variables clínicas y de laboratorios fueron la presencia de esplenomegalia, hepatomegalia, eventos trombóticos, hemorrágicos, cifras de hemoglobina, recuento de leucocitos y plaquetas, valores de lactato deshidrogenasa (LDH), el tipo de tratamiento empleado, la presencia de la mutación JAK2V617F, el riesgo trombótico, la respuesta al tratamiento y el tiempo de supervivencia global y libre de enfermedad.
Recolección, procesamiento y análisis de la información
Los datos se almacenaron en una base de datos confeccionada con el programa SPSS v.25.0 para Windows, a partir de la cual fueron procesados. Las variables cualitativas se resumieron a partir de sus frecuencias absolutas y relativas y para las variables cuantitativas la media y la desviación estándar.
El tiempo de supervivencia libre de enfermedad (SLE) se calculó desde la fecha en que ocurrió la remisión hematológica hasta la recaída del paciente y para la supervivencia global (SG) desde la fecha de diagnóstico hasta la fecha de la muerte.
El análisis de la supervivencia se realizó mediante el método de Kaplan-Meier y los resultados se compararon mediante la prueba de log-rank. Se consideraron significativos los valores de p≤ 0,05. Los resultados se presentan en tablas y gráficos.
Consideraciones éticas
Se presentó ante el Consejo Científico y el Comité de Ética de las Investigaciones el proyecto: “Caracterización de la trombocitemia esencial en pacientes adultos” que tiene como una de sus salidas la presente investigación. Se explicaron los objetivos, las técnicas a emplear y la importancia del estudio.
Se respetó lo establecido en los principios básicos de la Declaración de Helsinki que contiene las recomendaciones a seguir en la investigación biomédica con seres humanos.11 Se garantizó la confidencialidad de la información y la divulgación e introducción en la práctica de los resultados científicos que permiten la ampliación del conocimiento y el beneficio social a los pacientes.
Resultados
La trombocitemia esencial se presentó con mayor frecuencia en el grupo etario entre 39-58 años (55,0 %). El promedio de edad al diagnóstico fue 52,2 años y una mediana de 51 años. El 80 % se presentó en el sexo femenino y el 62,5 % de los pacientes de color de la piel blanca.
Tabla 1 Características sociodemográficas de los pacientes adultos con trombocitemia esencial
| Variable (n = 40) | No. | % |
|---|---|---|
| GRUPO ETARIO (AÑOS) | ||
| 18-38 | 5 | 12,5 |
| 39-58 | 22 | 55,0 |
| 59-78 | 12 | 30,0 |
| ≥ 79 | 1 | 2,5 |
| COLOR DE LA PIEL | ||
| blanco | 25 | 62,5 |
| no blanco | 15 | 37,5 |
| SEXO | ||
| femenino | 32 | 80,0 |
| masculino | 8 | 20,0 |
En relación con las características clínicas y de laboratorio al diagnóstico predominó los pacientes con comorbilidades (62,5 %). El 22,5 % de los casos presentó hepatomegalia y los eventos trombóticos se presentaron en un 12,5 % de los pacientes. Se observó una mayor frecuencia de los valores normales en las cifras de hemoglobina, hematocrito y conteo global de leucocitos, pero el 85 % de los pacientes presentaron conteo de plaquetas en el rango entre 450-1500 ( 109/L. El 52,1 % de los casos presentó aumento de la enzima lactato deshidrogenasa y la mutación del JAK2V617F representó el 67,5 %. La mayor parte de los pacientes se clasificaron en el grupo de riesgo intermedio según el índice internacional de trombosis (IPSET-trombosis), con un predominio de aquellos que presentaron respuesta estable.
El 72,5 % de los pacientes tuvo una enfermedad estable con el uso de las opciones terapéuticas de primera línea, y el 10 % presentó progresión de la enfermedad (tabla 2).
Tabla 2 - Relación del tratamiento de primera línea empleado con la respuesta al tratamiento de los pacientes adultos con trombocitemia esencial
| Respuesta al tratamiento | Aspirina | Hidroxiurea | Interferón convencional | Interferón pegilado | Total |
|---|---|---|---|---|---|
| No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | |
| Estable | 10 ( 90,9) | 13 (59,1) | 1 (100) | 5 (83,3) | 29 (72,5) |
| Refractaria | 1 (10,1) | 6 (27,3) | 0 (0) | 0 (0) | 7 (17,5) |
| Progresión | 0 (0) | 3 (13,6) | 0 (0) | 1 (16,7) | 4 (10) |
| Total | 11 (100) | 22 (100) | 1 (100) | 6 (100) | 40 (100) |
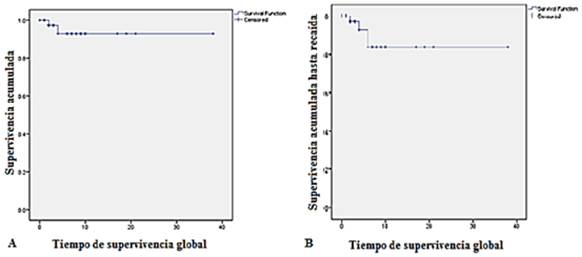
La probabilidad de la SG tuvo una media estimada en años de 35,5 con un IC 95 % (32,1-38,9) (A) y en cuanto a la probabilidad de la SLE, el análisis demostró una supervivencia estimada en años de 32,5 con un IC 95 % (27,5-35,5) (B) (fig.).
Discusión
La trombocitemia esencial se presenta con una edad media de 67 a 73 años y una mediana de 60 años, aunque un 20 % de los pacientes se diagnostican antes de los 40 años.12,13) Srour y otros plantean que se presenta una mediana de 55 años.14
En la investigación se demostró que la mediana de edad al diagnóstico, así como la media fue ligeramente inferior a lo que ellos reportan, aunque se considera que está acorde a lo revisado, en que es más frecuente su presentación a partir de la quinta década de la vida.
Además Srour y otros demostraron el predominio femenino en individuos menores de 60 años, así como un 18 % de incidencia mayor para los pacientes con color de la piel blanco.14 Estos resultados son semejantes a los que se reportan en la investigación.
El examen físico en los pacientes con TE es relativamente normal.2,3) Tefferi y otros reportan la incidencia de esplenomegalia hasta un 24 % y la hepatomegalia en el 20 %,4 resultados similares a los de esta serie. Sin embargo, cuando avanza la enfermedad puede aparecer un aumento adicional en el grado de hepatoesplenomegalia fundamentalmente en los que desarrollan una MF pos-TE.14,15
Los eventos trombóticos al momento del diagnóstico y durante el seguimiento ocurren en tasas del 10-29 % y del 8-31 %, respectivamente. Existen cifras variables de acuerdo a las series que difieren en otras características, como se constata en los estudios de Chim, Szubermy Srour.14,16,17) Los resultados coinciden con lo reportado en el estudio.
Los eventos hemorrágicos que se han descrito entre un 3-11 % de los pacientes con TE,2 no fueron encontrados en la serie estudiada, sin embargo, autores como Awada e Ianotto reportaron cifras de hasta un 23 % y 28 %, respectivamente.18,19
Investigaciones desarrolladas a partir de metanálisis han revelado que la mutación JAK2V617 se asocia con un riesgo dos veces mayor de desarrollar una trombosis arterial o venosa pero no influye en el riesgo de sufrir un evento hemorrágico.3,9,20
Otros estudios demuestran que un recuento de leucocitos elevado por encima de 11,0 ( 109/L se ha asociado a un mayor riesgo de desarrollar trombosis adicionales, que el grado de elevación del recuento de plaquetas.21,22 Estos informes contradictorios en la literatura entre expertos en este campo, hacen que sea cada vez más difícil ser dogmático sobre a quién tratar con agentes reductores de plaquetas y cuál debería ser el recuento de plaquetas objetivo.
El objetivo del tratamiento en la TE es prevenir eventos trombóticos y hemorrágicos adicionales sin aumentar el riesgo de transformación. La mayoría de los investigadores intentan normalizar el recuento de plaquetas o alcanzar uno en el que se resuelvan los síntomas del paciente de alto riesgo.23) Estos pacientes requieren reducción del número de plaquetas al rango normal con el uso de una variedad de agentes, que incluyen hidroxiurea, anagrelida o interferón α (IFNα).2
Las directrices actuales favorecen la hidroxiurea como tratamiento de primera línea en los pacientes que necesitan citorreducción.24
En el ensayo UK-PT1 en una cohorte de pacientes de alto riesgo diagnosticados según los criterios del PVSG,25) la recomendación se basa en la superioridad de la hidroxiurea combinada con ácido acetilsalicílico (ASA) frente a la anagrelida combinada con ASA. Generalmente la anagrelida no se considera como terapia de primera línea. El estudio ANAHYDRET demostró que la anagrelida no es inferior a la hidroxiurea en la reducción del riesgo trombótico en la TE de alto riesgo clasificada por la OMS.26
En la trombocitemia esencial, la ASA es de poco beneficio para la mayoría de los pacientes de bajo riesgo, con la excepción de los que presentan la mutación JAK2V617F, factores de riesgo cardiovascular (FRCV) o síntomas vasomotores.27) Los pacientes de alto riesgo deben recibir ASA, a menos que existan contraindicaciones. La citorreducción con hidroxiurea en combinación con ASA en dosis bajas, reduce el riesgo trombótico en los pacientes de alto riesgo.27,29
Otra terapia de primera línea es el IFNα, fármaco capaz de inducir reducciones de la carga alélica en el gen JAK2V617F en los pacientes con NMP.30) Un ensayo fase 2 de IFNα pegilado en PV y TE informó respuestas hematológicas y moleculares completas en el 70 % y 18 %, respectivamente.30) La exposición prolongada al IFNα pegilado es necesaria para lograr respuestas moleculares, mientras que las respuestas hematológicas se observan en los primeros 3 meses de terapia.27,28,29,30
Estos resultados concuerdan con lo encontrado en la investigación si se considera que todas las líneas de tratamiento utilizadas fueron efectivas en los pacientes estudiados con una respuesta hematológica estable de la enfermedad.
Según las recomendaciones del grupo europeo de leucemias (ELN por sus siglas en inglés), los pacientes se categorizan en: alto riesgo, si la edad > 60 años con historia de trombosis previas, y bajo riesgo para los pacientes con edad < 60 años y ausencia de trombosis.2,31)
Recientemente se utiliza el sistema de estratificación del riesgo trombótico (IPSET-trombosis por sus siglas en inglés),31 en el que se incluyen, además de los ya comentados, otros dos factores de riesgo adicionales, la mutación JAK2V617F y los FRCV. Los enfermos se categorizan en tres grupos (bajo, intermedio o alto riesgo según la puntuación sea menor, igual o mayor a 2); relacionado con este índice los pacientes mayores de 60 años, JAK2V617F negativos y sin factores de riesgo cardiovascular serían categorizados como de “bajo riesgo”, lo que tendría repercusión en la elección del tratamiento. El IPSET-trombosis, además, sitúa una elevada proporción de enfermos en la categoría de “riesgo intermedio”, en la cual no se ha analizado hasta la fecha ningún algoritmo de tratamiento. De acuerdo con este sistema pronóstico, el riesgo anual de trombosis para los grupos de riesgo bajo, intermedio y alto es de 1,03 %, 2,35 % y 3,56 %, respectivamente.2,31
Los resultados de esta investigación son similares, con lo referenciado por la literatura internacional al respecto, donde el riesgo intermedio fue el más frecuente en la población estudiada y los eventos trombóticos no fueron significativos.
Muchos autores refieren que el riesgo de progresión es extremadamente bajo en pacientes con TE no tratados. Estudios prospectivos más recientes tanto en TE como en PV han confirmado que la terapia con hidroxiurea se asocia con una baja incidencia de transformación leucémica cuando se usa sola (< 5 %) y con un seguimiento a largo plazo (≤ 14 años).25,32
En una serie con un seguimiento de aproximadamente 7 años, la mediana de supervivencia varió de 16 a 21 años, y la supervivencia de los pacientes con pre MF varió de 10,8 a 14,4 años, siendo la mayoría de las muertes atribuida a la progresión a MF manifiesta y LMA.33) La probabilidad de que un paciente con TE sobreviva 10 años varía entre el 64 % y el 80 %.33
Un estudio de 322 pacientes seguidos durante 13,6 años mostró un patrón diferente con una media de supervivencia de 35 años.17 En la presente serie, la media de supervivencia fue muy similar a las cifras reportadas por estos investigadores y superó al grupo de Luque Paz D y otros.33) Estos resultados se explican por el diagnóstico en edades más tempranas, la accesibilidad a los servicios de salud, el éxito de la estrategia terapéutica adaptada al riesgo de trombosis o pudiera estar influido por el número relativamente pequeño de casos estudiados.
Como conclusión los pacientes adultos con trombocitemia esencial en el IHI tienen una SG y SLE significativamente elevado, con baja incidencia de eventos trombóticos y el tratamiento empleado de primera línea se relacionó con la respuesta hematológica estable.

