










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Después de la susceptibilidad incrementada a las infecciones, la autoinmunidad representa el trastorno más frecuentemente asociado con las inmunodeficiencias primarias (IDP).1
Las inmunodeficiencias primarias conducen al funcionamiento aberrante del sistema inmune y predisponen a infecciones, inflamación/autoinmunidad, alergias y un riesgo elevado de padecer cáncer. Los síndromes por desregulación inmunológica que son IDP o se asocian a ellas, se evocan como una de las causas del vínculo entre IDP/enfermedad autoinmune (EAI).2
La prevalencia estimada de las IDP es de 1 en 1 200 pacientes en Estados Unidos de América.3) En Latinoamérica predominan las IDP humorales lideradas por el déficit selectivo de IgA, aunque ha existido un aumento progresivo en el reporte de IDP por desregulación.4 En Cuba, según datos del Registro Nacional de IDP, hasta el año 2017 existió un predominio de las deficiencias de anticuerpos (52,8 %).5
En el Registro Nacional de IDP de Francia se reportan manifestaciones autoinmunes e inflamatorias en el 26,2 % de los pacientes.6 En Cuba en el 2017 se reportó que el 28,13 % de los niños inmunodeficientes atendidos en La Habana presentaron diagnóstico de enfermedad autoinmune, a diferencia de solo 3 pacientes (4,69 %) del grupo control.7
La descripción de las manifestaciones y las enfermedades autoinmunes en poblaciones de pacientes con IDP es útil y sienta las bases para futuras investigaciones. Puede contribuir a acortar el tiempo de diagnóstico de las IDP. Ayuda a la realización de un diagnóstico de las mismas en etapas subclínicas, sobre todo si se tiene en cuenta que los fenómenos autoinmunes son el segundo hallazgo más frecuentemente descrito en pacientes con IDP. Pueden ser la manifestación inicial de una IDP tanto en niños como en adultos y en los primeros como una condición grave.
Administrar tratamientos inmunosupresores a pacientes con manifestaciones o enfermedades autoinmunes sin tener en cuenta que pueden ser el indicio de una IDP puede llevar a descompensaciones fatales de esta última, por lo que se hace necesario conocer y luego precisar en cada paciente la existencia de esta relación. Finalmente, la investigación discutió aspectos que cambian positivamente la visión de la comunidad médica para el diagnóstico de las IDP. Por ello el objetivo fue determinar el comportamiento de las enfermedades autoinmunes en los pacientes con inmunodeficiencias primarias.
Métodos
Se realizó un estudio analítico, de tipo casos y controles, en el Centro Médico Ambulatorio del Hospital Provincial Carlos Manuel de Céspedes de Bayamo, Granma, en el período comprendido entre enero de 2013 y enero de 2022.
El universo del estudio estuvo conformado por pacientes que habían recibido atención médica especializada por Inmunología y que tuvieran recogido en su historia clínica los datos necesarios para la investigación.
La muestra fue de 114 pacientes separados en dos grupos. El grupo de casos: constituido por los 38 pacientes con IDP diagnosticadas, confirmadas, clasificadas y valorados por reumatología. El grupo control lo constituyó 76 pacientes. Para el “control” de los factores de confusión, por cada paciente con diagnóstico de IDP se parearon dos pacientes sin compromiso del sistema inmune que se seleccionaron por muestreo aleatorio simple entre los miembros del universo que tenían los datos completos en las historias clínicas y que habían sido valorados por reumatología.
Se analizaron como variables la edad y el tipo de IDP. La edad se separó en dos grupos; 18 o menos años y mayores de esta edad. El tipo de IDP se estableció según la clasificación de la IUIS (del inglés, International Union of Immunological Societies) del 2020. El diagnóstico de las enfermedades autoinmunes se realizó en consulta de Reumatología según los criterios vigentes para cada una de las enfermedades, la presencia de manifestaciones sugerentes autoinmunidad se tomó de los datos recogidos en las historias clínicas.
El análisis de los datos se realizó en el programa estadístico SPSS 25. Se emplearon frecuencias absolutas y relativas. Se realizó un análisis univariado y se evaluaron los odds ratio.
Se cumplió con los principios éticos de la investigación científica.
Resultados
Las manifestaciones clínicas sugerentes de autoinmunidad aparecieron en 15 de los pacientes inmunodeficientes (83,33 %); sin embargo, solo 16,67 % de los pacientes con competencia inmunológica presentaron síntomas sugerentes de autoinmunidad (OR 15,869; p= 0,000).
Las manifestaciones sugestivas de autoinmunidad, al analizarlas por grupos de edades se comportaron de la misma manera. De los 38 pacientes que formaron el grupo de estudio 25 tenían 18 o menos años y 13 eran mayores de esa edad. En el grupo de 18 o menos, de 7 pacientes con manifestaciones sugerentes de autoinmunidad 6 son pacientes con inmunodeficiencia (85,7 %) y en los de más de 18,9 de los pacientes con autoinmunidad (81,8 %) tenían algún tipo de inmunodeficiencia de base (tabla 1).
Tabla 1 Presencia de manifestaciones sugerentes de autoinmunidad en pacientes inmunodeficientes según grupos de edades
| Síntomas sugerentes de autoinmunidad |
Edad ≤ 18 años, 11 meses, 29 días [n(%)] |
Edad ( 18 años [n(%)] |
Total [n(%)] |
|||
|---|---|---|---|---|---|---|
| IDP | NO IDP | IDP | NO IDP | IDP | NO IDP | |
| Presentes | 6 (85,7) | 1 (14,3) | 9 (81,8) | 2 (18,18) | 15 (83,33) | 3 (16,67) |
| Ausentes | 19 (27,9) | 49 (72,1) | 4 (14,3) | 24 (85,71) | 23 (23,96) | 73 (76,04) |
| OR | 27,000 | 11,000 | 15,869 | |||
| 0,0001 | 0,0027 | 0,000 | ||||
IDP: inmunodeficiencia primaria; OR: odds ratio.
Las manifestaciones de autoinmunidad en los pacientes con IDP fueron, en orden de frecuencia: dolor monoarticular en 6 pacientes (33,33 %), poliartralgia, dermatitis y alopecia con 3 casos (16,67 %) cada uno, seguidos por dolor muscular, dolor en columna vertebral y nódulos subcutáneos presentes en un paciente cada uno (5,56 %).
El 73,33 % de los niños con manifestaciones clínicas sugerentes de autoinmunidad tenían diagnóstico de IDP predominante de anticuerpos que incluyeron los diagnósticos de déficit de IgA, déficit de IgG e inmunodeficiencia variable común (IDVC). El análisis de prevalencia individual de cada tipo de IDP por separado arrojó que el 42,30 % de los pacientes con diagnóstico de IDP predominantemente de anticuerpos presentó algunas de las manifestaciones estudiadas. El 100 % de los pacientes con inmunodeficiencia por desregulación y con síndrome de DiGeorge, presentaron alguna manifestación sugerente de autoinmunidad (tabla 2).
Tabla 2 Presencia de manifestaciones sugerentes de autoinmunidad según tipos de inmunodeficiencias primarias
| Tipos de IDP | Presencia de manifestaciones sugerentes de autoinmunidad | Sin manifestaciones sugerentes de autoinmunidad | Total | |||
|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |
| Agammaglobulinemia ligada al X | 0 | 0 | 1 | 100 | 1 | 100 |
| Candidiasis mucocutánea | 0 | 0 | 1 | 100 | 1 | 100 |
| IPEX- |
1 | 100 | 0 | 0 | 1 | 100 |
| Inmunodeficiencia variable común | 4 | 66,67 | 2 | 33,33 | 6 | 100 |
| Neutropenias | 0 | 0 | 3 | 100 | 3 | 100 |
| Hipoplasia tímica con inmunodeficiencia clínica | 2 | 40 | 3 | 60 | 5 | 100 |
| Síndrome de DiGeorge | 1 | 100 | 0 | 0 | 1 | 100 |
| Déficit de IgA | 4 | 26,67 | 11 | 73,33 | 15 | 100 |
| Déficit de IgG | 3 | 60 | 2 | 40 | 5 | 100 |
En el grupo de casos 13 pacientes (72,22 %) presentaron diagnóstico de enfermedad autoinmune, a diferencia de 5 pacientes (27,78 %) del grupo control, con p= 0,0011 y OR; IC 95 % de 7,384. Las enfermedades autoinmunes diagnosticadas en pacientes con IDP fueron en orden de frecuencia: enfermedad celiaca en 4 (30,71 %), vitíligo 3 (23,07 %), fibromialgia 2 (15,38 %); seguidos con igual frecuencia del eritema nodoso, la gastritis eosinofílica, la anemia perniciosa y la vasculitis (púrpura de Schönlein-Henoch) con un paciente por cada diagnóstico (7,69 %).
De los menores de 18 años presentaron autoinmunidad 6 pacientes inmunodeficientes (75 %) y 2 (25 %) de los inmunocompetentes. En el grupo de mayores de 18 años la autoinmunidad apareció en el 70 % de los pacientes con IDP y en 3 pacientes sin IDP (tabla 3).
Tabla 3 Diagnóstico de enfermedades autoinmunes en pacientes con inmunodeficiencias primarias (IDP) según grupos de edades
| Diagnóstico de autoinmunidad |
Edad < 18 años, 11 meses, 29 días [n(%)] |
Edad ( 18 años [n(%)] |
Total [n(%)] |
|||
|---|---|---|---|---|---|---|
| IDP | Sin IDP | IDP | Sin IDP | IDP | Sin IDP | |
| Presentes | 6 (75) | 2 (25) | 7 (70) | 3 (30) | 13 (72,22) | 5 (27,78) |
| Ausentes | 19 (28,36) | 48 (61,34) | 6 (20,69) | 23 (69,31) | 25 (26,02) | 71 (73,96) |
| OR | 27,000 | 8,944 | 7,384 | |||
| 0,0001 | 0,0043 | 0,0001 | ||||
Los pacientes con agammaglobulinemia ligada al cromosoma X, neutropenias primarias, síndrome de DiGeorge, hipoplasia tímica con manifestaciones de inmunodeficiencia y candidiasis mucocutánea crónica, no presentaron enfermedades autoinmunes. El paciente con IPEX-like presentó enfermedad celiaca, al igual que 3 de los pacientes IDVC. En esta última, además de celiaquía, se reportó anemia perniciosa en un paciente. En los tres pacientes con déficit de IgA, se diagnosticó vitíligo, gastritis eosinofilicas y vasculitis, uno en cada caso y en el déficit de IgG apareció eritema nodoso en un caso y fibromialgia en dos (fig.).
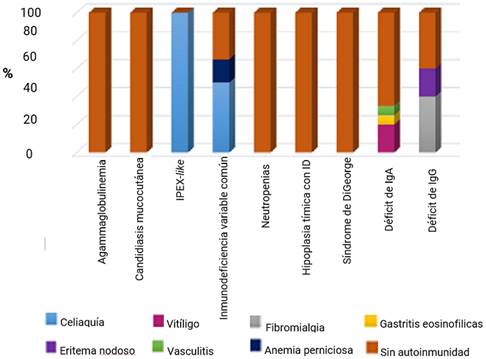
Discusión
Se han planteados varios mecanismos para explicar la paradójica ocurrencia de enfermedades autoinmunes durante las IDP, incluyendo una anomalía en la selección negativa de las células T autorreactivas en el timo, una deficiencia cualitativa o funcional de los linfocitos T reguladores, una alteración de la apoptosis de los linfocitos autorreactivos y una pérdida de tolerancia central o periférica.2
Las enfermedades autoinmunes pueden ser la manifestación inicial de una IDP, tanto en niños como en adultos, pero en los primeros como una condición severa y en los segundos con mayor frecuencia.8
Las enfermedades autoinmunes debutan frecuentemente de manera progresiva y solapada. Comienzan con surgir manifestaciones aisladas que evolucionan o van sumándose a lo largo de los años y puede o no aparecer la enfermedad autoinmune. Las manifestaciones de esta índole son hoy el segundo grupo de síntomas y signos asociados a IDP, solo superado por las infecciones. Por estas razones se estudió tanto las manifestaciones sugerentes de autoinmunidad como las EAI y se separaron por grupos de edades.
En el Registro Nacional de IDP de Francia se encontraron manifestaciones autoinmunes e inflamatorias en el 26,2 % de los pacientes con gran riesgo de aparecer durante sus vidas.6 La presente investigación muestra mayor incidencia de manifestaciones autoinmunes que en Francia.
En Cuba en el año 2017 se constató en niños con IDP una mayor incidencia de manifestaciones sugerentes de autoinmunidad (46,87 %) que el grupo control, en el que solo se reportó un 7,18 %.7 La presente investigación coincide con esos resultados. El estudio cubano informó que entre las manifestaciones sugerentes de autoinmunidad se encontraban las artralgias o artritis (40 %). Las citopenias no relacionadas con el diagnóstico primario y las dermatitis tuvieron igual prevalencia, siendo reportadas cada una 26,6 %.7 Los resultados expuestos están acordes con estos datos pues tres manifestaciones se encontraron en los pacientes estudiados y solo no se describieron las citopenias.
En la literatura revisada, la dermatitis eccematosa apareció en pacientes con inmunodeficiencia combinada severa (SCID) y síndrome de poliendocrinopatía desregulación y enteropatía ligada al cromosoma X (IPEX) y síndrome de Wiskott Aldrich (WAS), la anemia hemolítica, la trombocitopenia y las neutropenias aparecieron en pacientes con déficit de IgA, IDVC y defectos de cambios de clase.9
En la literatura internacional la alopecia se asoció al síndrome de Omenn, al síndrome de poli-endocrinopatía con distrofia ectodérmica y candidiasis (APECED). Los síndromes IPEX e IPEX-like y en menor medida al déficit de IgA y las artralgias de 1 a 10 % en IDVC, 4 % en déficit de IgA.10 La presente investigación coincidió parcialmente con estos reportes.
Cuatro de los pacientes con IDVC tenían manifestaciones e incluso estudios citométricos que permiten plantear que pertenecen al grupo de pacientes en las que la IDCV concomita con desregulación. En los resultados se mostraron todos los pacientes con este diagnóstico como inmunodeficientes con predominio de anticuerpos, pues oficialmente por la clasificación internacional pertenecen a este grupo. Se pretende hacer notar que, al analizar estos 4 casos como enfermedades desregulatorias serían 5 de 7 casos con inmunodeficiencia por desregulación que presentarían EAI, lo que constituye el 71,43 % y se corresponde con la literatura internacional que ubica a las desregulación en una asociación cercana al 80 % con autoinmunidad.6
Odnoletkova y otros, en 2018 plantearon que pacientes con IDVC pueden mostrar un fenotipo similar a IPEX y ellos pueden presentar citopenias, enfermedad inflamatoria del intestino, alergias, eczema, granulomas, linfoproliferación o malignidades.9 Los resultados están en concordancia con los de Laura Berrón-Ruiz que planteó que en la IDCV hasta el 25 % de los pacientes presenta manifestaciones autoinmunitarias y que dada la frecuencia y la aparición temprana en algunos pacientes con IDCV, la desregulación de las enfermedades autoinmunitarias parece ser una parte integral de esta inmunodeficiencia.11 En este estudio no se reportaron citopenias diferentes del diagnóstico primario. Otras manifestaciones autoinmunes en pacientes con IDVC incluyen artralgia, artritis, alopecia, entre otras.1
En niños cubanos inmunodeficientes, el 86,7 % de los que presentaron manifestaciones clínicas sugerentes de autoinmunidad pertenecían a las IDP predominantemente de anticuerpos.7 La presente investigación coincide con esos resultados. Sin embargo, en ese estudio reportaron manifestaciones autoinmunes en pacientes con déficit de complemento e inmunodeficiencia combinada, respectivamente7 a diferencia de lo encontrado en el presente estudio ya que no se reportó este tipo de IDP.
La presente investigación reporta un paciente con inmunodeficiencia por desregulación (IPEX like), alopecia y dermatitis eccematosa, lo que coincide con otras naciones que mostraron en enfermos de IPEX la aparición de lesiones en piel entre un 42 y 60 %.12) La alopecia también se asoció en la literatura con síndromes desregulatorios,13 con IDVC y con APECED.10) Entre los pacientes con diagnóstico de IPEX estudiados en otras latitudes, el 92 % presentó enteropatía.14)
Ante la dificultad diagnóstica para diferenciar las alteraciones en las biopsias típicas de la desregulación y de la enfermedad celiaca se emplearon otros medios como los marcadores genéticos y los autoanticuerpos típicos de la enfermedad celiaca, así como los resultados de la dieta de exclusión.
En Cuba, en el año 2017, se reportó que el 28,13 % de los niños inmunodeficientes atendidos en La Habana presentaron diagnóstico de enfermedad autoinmune, a diferencia de solo 3 pacientes (4,69 %) del grupo control.7 Se coincide con el predominio de diagnóstico autoinmune en IDP.
Sin embargo, el citado estudio cubano reportó enfermedades autoinmunes en orden de frecuencia con las que no concordamos en su totalidad,7 porque en la presente investigación no se registró paciente con lupus eritematoso sistémico (LES), ni artritis idiopática juvenil.
En la investigación que se comparte en este artículo también se encontró asociación de IDP desregulatoria en un elevado por ciento con enfermedades autoinmune, lo que destaca su vínculo con la enfermedad celiaca tanto del caso de IPEX-like como en 2 de las 4 IDVC que se asocian con desregulación. Estos aspectos también coinciden con Caldirola y otros, que en el año 2020 reportaron enfermedades autoinmunes asociadas en más del 90 % con IPEX13 y enfermedad celiaca en el 4 % de los pacientes con IDVC.15
Los resultados del estudio coinciden con reportes internacionales de aparición de vitíligo y anemia perniciosa en pacientes con IDP.11 Se describió en el caso del déficit de IgA la asociación con vitíligo, gastritis eosinofílica y vasculitis (púrpura de Schönlein-Henoch). La literatura revisada en el caso del déficit de IgA, reporta asociación en aproximadamente del 20 al 30 % con púrpura trombocitopénica idiopática seguida de anemia hemolítica, artritis reumatoide juvenil, vitíligo, tiroiditis, vasculitis, lupus eritematoso sistémico y presencia de diversos autoanticuerpos.10,16
En España, de 330 pacientes menores de 18 años con déficit de IgA, 22 (6,6 %) eran celiacos y 38 (11,5 %) presentaron enfermedades autoinmunes (artritis crónica juvenil, diabetes mellitus, vitíligo, citopenias y enfermedad de Crohn).17) La presente investigación mostró (en el caso particular de la IDVC) su asociación con la enfermedad celiaca y anemia perniciosa.
En la IDVC un subgrupo de pacientes se caracteriza por manifestaciones adicionales, a menudo predominantes, de desregulación inmunitaria.11 Aproximadamente, un 30 % de los pacientes con IDCV desarrollan enfermedades autoinmunes. La mitad de las complicaciones se pueden atribuir a citopenia autoinmunitaria.10,11) Otras enfermedades autoinmunes en pacientes con IDVC incluyen la anemia perniciosa, el síndrome de Sjögren, uveítis, vasculitis, tiroiditis, alopecia, vitíligo, hepatitis, cirrosis biliar primaria, síndrome Zika o LES.10,15,18
Además, los pacientes con IDVC y enteropatía pueden presentar manifestaciones similares a las de la enfermedad celiaca y enfermedad celiaca como tal.11) La presente investigación muestra un paciente con inmunodeficiencia por desregulación que padece de enfermedad celiaca.
Los pacientes con agammaglobulinemia ligada al cromosoma X, neutropenias, síndrome de DiGeorge, hipoplasia tímica con manifestaciones de inmunodeficiencia y candidiasis mucocutánea crónica no presentaron enfermedades autoinmunes. En estos aspectos se encontró discrepancia con la literatura que mostró que las enfermedades autoinmunes pueden ser parte del síndrome de DiGeoerge19 y que la gammaglobulinemia se asocia con menos de un 20 % con autoinmunidad.20
Las manifestaciones y enfermedades autoinmunes prevalecieron en pacientes con IDP en relación con pacientes sin compromiso inmunológico, con mayor frecuencia en los mayores de 18 años. Las inmunodeficiencias más frecuentes asociadas a los trastornos autoinmunes fueron las inmunodeficiencias de anticuerpos y las que tenían algún componente de desregulación.
Las manifestaciones sugerentes de autoinmunidad con mayor presencia fueron: dolor monoarticular, poliartralgia, dermatitis, dolor muscular, dolor en columna vertebral y nódulos subcutáneos. La enfermedad celiaca, vitíligo, fibromialgia, eritema nodoso, gastritis eosinofílicas, anemia perniciosa y la vasculitis (púrpura de Schönlein-Henoch) fueron las enfermedades autoinmunes más frecuentemente diagnosticadas.

