










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
La enfermedad de Gaucher (EG) (OMIM 230806) es una enfermedad de depósito lisosomal caracterizada por un defecto en el metabolismo de los glucocerebrósidos, producida por la disminución en la actividad de la enzima lisosomal β glucocerebrosidasa ácida. 1
Fue descrita inicialmente por Philippe Charles Ernest Gaucher, en 1881.2) Posteriormente, Brill y cols.3 describieron su forma de herencia autosómica recesiva y, en 1965, Braddy y cols. identificaron la actividad deficiente de la enzima.4
Presenta una incidencia de 1/40 000 a 1/60 000 nacimientos en la población general, la cual asciende a 1/800 nacidos vivos en la población de judíos de Ashkenazi.5
El defecto enzimático provoca el acúmulo del sustrato natural de la enzima, la glucosilceramida, que actúa como un potente estímulo activador del macrófago y provoca una serie de alteraciones secundarias del metabolismo lipídico y niveles de proteína en el plasma y en el líquido cefalorraquídeo.6
El gen que codifica la enzima se encuentra en el brazo largo del cromosoma1(1q21), contiene once exones y presenta un pseudogen con alta homología (96 %) que puede ocasionar dificultades en la determinación de los genotipos. Están descritas más de 500 mutaciones, que pueden ser de tipo sin sentido (las más frecuentes), deleciones, inserciones y por alelos recombinantes. Entre las mutaciones más frecuentes se encuentran: N370S, L444P,84GG, IVS2(+ 1) y D409H.6
La enfermedad se caracteriza por su variabilidad clínica, que implica la diversidad en la sintomatología, tanto al inicio de las manifestaciones clínicas como en su intensidad. Están descritos tres tipos clásicos: el tipo 1 no neuronopático, el más frecuente (94 %); el tipo 2 o neuronopático agudo (1 %) y el tipo 3 neuronopático crónico (5 %); los dos últimos con afectación del sistema nervioso central. A pesar, de que la clasificación es útil para el pronóstico y el tratamiento, en la actualidad se plantea la existencia de un fenotipo continuo, más que una categorización separada de los clásicos tipos de la enfermedad.7)
Clínicamente se caracteriza por esplenomegalia, hepatomegalia, anemia, trombocitopenia y alteraciones esqueléticas que conducen a numerosas complicaciones óseas como: deformidad en matraz de Erlenmeyer, osteopenia, osteosclerosis, osteonecrosis, lesiones líticas, fracturas patológicas y crisis óseas. Las principales manifestaciones neurológicas son: apraxia oculomotora, estrabismo, espasticidad, retroflexión de la cabeza, hiperreflexia, trismus, disfagia, estridor laríngeo, alteraciones extrapiramidales, crisis convulsivas, mioclonias. En el tipo 2 estas manifestaciones se presentan y evolucionan de forma más rápida y tórpida, lo que conduce a la muerte de los pacientes antes de los dos años de edad.
El diagnóstico molecular de la EG se realiza, ante la sospecha clínica, por la historia familiar, historia clínica, hallazgos radiológicos y la determinación de la actividad enzimática de la β-glucocerebrosidasa.8
La terapia de reemplazo enzimático (TRE) ha modificado la historia natural de la enfermedad, desde su aprobación por la Administración de Drogas y Alimentos (FDA, por sus siglas en inglés), en 1991. Inicialmente fue producida a partir de placentas humanas y posteriormente obtenida a partir de tecnología de ADN recombinante, es conocida como imiglucerasa y comercializada como Cerezyme®. La TRE mejora los parámetros hematológicos y las visceromegalias, con la desventaja que la enzima recombinante no atraviesa la barrera hematoencefálica, por lo que las manifestaciones neurológicas se mantienen.9
Este trabajo tiene como objetivo evaluar la respuesta al tratamiento sustitutivo enzimático con imiglucerasa en pacientes cubanos con enfermedad de Gaucher.
Método
Se realizó un estudio longitudinal, descriptivo en el que se evaluó el comportamiento de variables clínicas, hematológicas y ultrasonográficas de ocho pacientes con TRE con imiglucerasa; al año, a los cinco y de diez a quince años de tratamiento. Se estudiaron las cifras de hemoglobina, de plaquetas, el tamaño del bazo e hígado por ultrasonido. Se utilizaron los resultados de los estudios recogidos en las historias clínicas del Instituto de Hematología e Inmunología, sin revelar datos personales de los participantes.
En la actualidad en Cuba hay once pacientes con TRE, apoyados para su tratamiento por el Programa Humanitario de Sanofi. Para el estudio solo se tomaron ocho, pues los tres restantes llevan seis meses o menos de tratamiento.
Resultados
De los ocho pacientes con TRE, seis adultos y dos en edad pediátrica, la mayoría fueron diagnosticados antes de los seis años y tenían más de 10 años con el TRE (imiglucerasa).
Todos los pacientes presentaron anemia al inicio y la mayoría tuvieron trombocitopenia y hepatoesplenomegalia al diagnóstico de la enfermedad. Los pacientes con manifestaciones neurológicas y la mutación L444P en estado homocigótico se clasificaron en EG tipo3, el resto en tipo1. (tabla 1)
Tabla 1. Variables demográficas, manifestaciones clínicas al diagnóstico, genotipo y tipo de enfermedad de Gaucher
| Paciente | Edad (años) | Manifestaciones clínicas | Genotipo | Años de TRE | Tipo | ||||
|---|---|---|---|---|---|---|---|---|---|
| Actual | Al diagnóstico | Anemia/Trombocitopenia | Visceromegalia | Óseas | Neurológicas | ||||
| 1 | 21 | 4 | ++ | +++ | - | - | N370S/I403T | 16 | I |
| 2 | 17 | 2 | +++ | +++ | + | ++ | L444P/L444P | 16 | III |
| 3 | 18 | 3 | +++ | + (No hepatomegalia) | ++ | + | L444P/L444P | 16 | III |
| 4 | 13 | 1 | +++ | +++ | ++ | +++ | L444P/L444P | 12 | III |
| 5 | 23 | 17 | +++ | +++ | + | - | N227S/ ? | 6 | III |
| 6 | 39 | 6 | +++ | +++ | - | N370S/N370S | 20 | I | |
| 7 | 61 | 7 | +++ | +++ | - | - | N370S/L444P | 20 | I |
| 8 | 27 | 6 | +++ | +++ | ++ | ++ | L444P/? | 20 | III |
TRE: terapia de reemplazo enzimático con imiglucerasa
En la tabla 2 se muestra la evolución del tamaño del bazo y el hígado. En todos los pacientes se constató esplenomegalia previa al tratamiento. Al año de la TRE, hubo respuesta favorable con disminución del tamaño del bazo en el 100 % de los pacientes; al igual que en el tamaño del hígado. Solo un paciente se mantiene con ligera esplenomegalia residual.
Tabla 2 . Evaluación del tamaño del bazo e hígado (cm) durante la terapia de reemplazo enzimático (TRE)
| Pacientes | Bazo (cm) | Hígado (cm) | ||||||
| Previo | 1 año | 5 años | 10-15 años | Previo | 1 año | 5 años | 10-15 años | |
| 1 | Accesorio (6,3) | 4 | 3,5 | Normal | + 6 | + 2 | + 2 | Normal |
| 2 | Masiva | 6 | 6,5 | Normal | + 5 | + 2 | Normal | Normal |
| 3 | 13,2 | 9,1 | 9,3 | Normal | Normal | Normal | Normal | Normal |
| 4 | Masiva | 6,9 | 6,9 | Normal | + 4 | + 3 | Normal | Normal |
| 5 | Masiva | 19,5 | 10,5 | Normal | + 12 | + 2 | Normal | |
| 6 | 15 | 11,5 | 12 | Normal | + 4 | Normal | Normal | Normal |
| 7 | 15 | 12 | 13 | Ligera esplenomegalia | + 4 | Normal | Normal | Normal |
| 8 | Masiva | 9 | 7 | Normal | + 5 | + 3 | Normal | Normal |
Medidas en cm que rebasan el reborde costal, realizado por ultrasonido
Con relación a las variables hematológicas, en la figura 1 se muestra el comportamiento de las cifras de hemoglobina previo, después de 1 año, a los 5 años y 10 años de tratamiento. El 100 % de los pacientes presentaban anemia antes del tratamiento, luego de un año con la terapia se alcanzaron y mantuvieron cifras normales de hemoglobina en todos los pacientes.
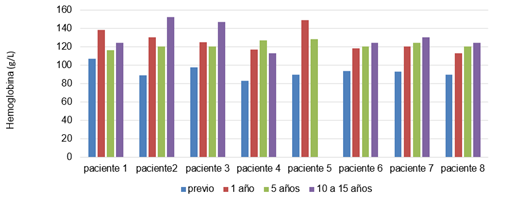
Fig. 1 - Cifras de hemoglobina de los pacientes con enfermedad de Gaucher durante el tratamiento con Imiglucerasa.
En la figura 2 se muestra el conteo plaquetario previo y durante el tratamiento; la mayoría de los pacientes presentaban cifras de 150 x 109 o trombocitopenia en el momento del diagnóstico, las que ascendieron tras 1 año de administración de la imiglucerasa, excepto el paciente 7 que se mantiene con cifras plaquetarias en el límite inferior.
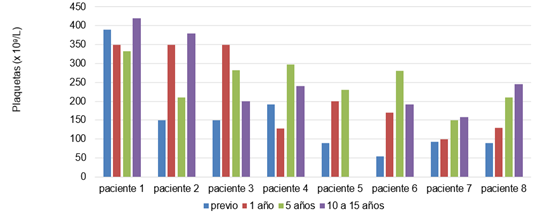
Discusión
La EG presenta gran variabilidad clínica y molecular, en particular el tipo 1 puede presentar el inicio de sus síntomas a cualquier edad. En el Registro de Gaucher regido por un Grupo Colaborativo Internacional, el 56 % de los pacientes fueron diagnosticados antes de los 20 años.8 Los pacientes del estudio presentaron los síntomas en edades tempranas. En el caso del paciente 5, aunque el diagnóstico fue a los 18 años, tuvo manifestaciones óseas en la edad pediátrica que se interpretó como un cuadro de enfermedad de Perthes.
Los signos y síntomas al momento del diagnóstico se caracterizan por anemia, generalmente moderada (20-50 %); trombocitopenia (60-90 %); esplenomegalia, en ocasiones masiva (más del 90 %); hepatomegalia (60-80 %) y manifestaciones óseas entre los más frecuentes. Estas manifestaciones clínicas se presentaron en todos los pacientes estudiados al inicio de la enfermedad, siendo la esplenomegalia masiva un signo recurrente en la mayoría de ellos. El compromiso neurológico, fundamentalmente en el tipo 3, es muy variable; algunos pacientes solo presentan oftalmoplejía horizontal (paciente 3) y otros casos expresiones más graves que incluyen epilepsia mioclónica progresiva, ataxia cerebelar, espasticidad y demencia (paciente 4).10,11) En ocasiones los signos neurológicos pueden aparecer años después de las manifestaciones viscerales, o estar presentes de forma muy ligeras y no ser detectadas al examen físico neurológico, por lo que inicialmente el paciente puede ser clasificado como una EG tipo1 y en su revaluación reclasificado como tipo 3; situación que sucedió con la paciente 5 del estudio.
Están descritas más de 500 mutaciones relacionadas con la EG entre ellas las más frecuentes reportadas se encuentran la N370S, L444P, IVS2+1G>A, lo cual coincide con la mayoría de las mutaciones detectadas en los pacientes del estudio.11 La presencia de la N370S en estado homocigótico o heterocigótico excluye el riesgo de afectación neurológica, aunque se reporta un incremento en el riesgo de desarrollar enfermedad de Parkinson.12,13) Sin embargo, los pacientes homocigóticos para L444P presentan un alto riesgo de desarrollar manifestaciones neurológicas. A pesar de lo descrito, aún la correlación fenotipo-genotipo para predecir el curso de la enfermedad se torna difícil. Así, se observan pacientes con igual estado mutacional (pacientes 2, 3 y 4) con la mutación en homocigosis (L444P/L444P), pero con diferentes comportamientos clínicos y respuesta al tratamiento, por lo que se plantean otros posibles modificadores genéticos que influye en el fenotipo de la EG.14,15
El TRE revolucionó el tratamiento de la enfermedad y marcó un beneficio para el pronóstico de los pacientes con EG tipo 1 y 3, pues no está indicada en el tipo 2. Aunque las dosis de imiglucerasa pueden variar, se recomienda comenzar con dosis de 60 u/kg cada 15 días, por vía endovenosa. El ajuste de la dosis de mantenimiento se realiza de forma individual y se disminuye o aumenta según el logro de las metas terapéuticas evaluadas en el monitoreo de cada paciente. Aunque no tiene impacto en las manifestaciones neurológicas por no atravesar la barrera hematoencefálica, se obtiene una gran mejoría de los parámetros hematológicos, viscerales y de la calidad de vida de los pacientes. (8 La falta de respuesta en seis meses indica que el paciente necesita una dosis más alta. La mayoría de los pacientes presentados en el trabajo alcanzaron sus metas terapéuticas en solo un año de tratamiento con imiglucerasa. En ocasiones la trombocitopenia puede mantenerse en pacientes con una esplenomegalia residual, como el paciente 7 del estudio.
La seguridad del medicamento es muy alta, solo del 2 al 4 % de los pacientes desarrollan anticuerpos contra la enzima, usualmente sin signos clínicos. Las reacciones alérgicas aparecen en menos del 1,5 % de los pacientes, 16 lo cual no se reportó en ninguno de los casos del estudio. No está contraindicado durante la gestación, ni relacionado con malformaciones fetales, de hecho la paciente 6 continuó tratamiento durante el embarazo sin complicaciones. 17
La TRE con imiglucerasa constituye una opción de tratamiento importante para los pacientes con EG tipo 1 y tipo 3. Su comienzo en edades pediátricas permite una evolución favorable de los pacientes, con alcance y cumplimiento de las metas terapéuticas, así como un mejoramiento en su calidad de vida. La interrupción se asocia con recaída de los parámetros hematológicos y esqueléticos, así como de los volúmenes de los órganos.

