










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versão impressa ISSN 0864-0300versão On-line ISSN 1561-3011
Rev Cubana Invest Bioméd v.16 n.2 Ciudad de la Habana jul-dez. 1997
Participación plaquetaria en la hemostasia primaria
Dr. Andrés O. Pérez Ruíz, Dr. José A. Castillo Herrera, Dra. Teresa Gortazar González, Dr. Miguel Alvarez Fornari, Dr. Roberto Douglas Pedroso y Dra. Belsys Díaz RondónLas plaquetas son discos de forma elipsoidal que como todas las demás células sanguíneas provienen de una célula hematopoyética Stem, fuente o tronco multipotente que da lugar a la línea megacariocítica. En 1906, Wright demostró que estas partículas se formaban en la médula ósea por la fragmentación del citoplasma de una célula poliploide, el megacariocito.
El recuento normal de plaquetas es de 150-350 x 109/L y tienen como función principal la de prevención y detención de la pérdida de sangre del torrente circulatorio.
Se calcula que en un individuo normal, 42 x 109 plaquetas/L/día aproximadamente se renuevan de la circulación, con un tiempo promedio de supervivencia de 8 días, siendo el bazo el sitio donde se destruyen ya que un tercio de las plaquetas en circulación se localizan en él debido al enlentecimiento de la corriente sanguínea en los sinusoides esplénicos.
Para la comprensión del fenómeno de adhesión y agregación plaquetaria, así como de la liberación de sus gránulos es importante comprender las características estructurales especiales de las plaquetas.
CITOESQUELETO PLAQUETARIO
El citoesqueleto plaquetario es el responsable de mantener la estabilidad de la membrana, su forma discoide y las modificaciones morfológicas que éstas experimentan cuando son activadas. En él pueden distinguirse 3 importantes estructuras:
- El esqueleto citoplasmático. Contiene una alta proporción de actina, la proteína más abundante en la plaqueta. En la plaqueta en reposo esta actina se encuentra formando filamentos que tienen uniones cruzadas con otras proteínas, lo que condiciona la formación de una malla tridimensional que funciona como un esqueleto celular. De esta manera, la actina se localiza en la zona de formación de pseudópodos, pudiendo unirse a la porción intramembranosa del complejo glicoprotéico IIb-IIIa y promover líneas de tensión y favorecer la retracción del coágulo.
- El esqueleto de membrana. Está formado también por filamentos de actina y se encuentra dispuesto inmediatamente por debajo de la membrana celular. Este sistema es el responsable de mantener la estabilidad de la membrana y capacita al complejo glicoproteico Ib-IX en su unión con el factor de Von Willebrand, propiciando la adhesión de la plaqueta al subendotelio.
- El anillo de microtúbulos. Está situado inmediatamente por debajo del esqueleto de la membrana y su función principal estriba en mantener la forma discoide de las plaquetas en estado basal y además centran los gránulos de la plaqueta cuando ésta es activada y su consecuente liberación por un proceso de exocitosis.
GRÁNULOS
Varios tipos de gránulos se observan en el citoplasma plaquetario: las mitocondrias, que capacitan a estas estructuras en la oxidación de ácidos grasos proveyéndolas de metabolismo aeróbico; los cuerpos densos que son reservorios de serotonina, nucleótidos adenílicos metabólicamente no utilizables por la plaqueta e iones calcio y los gránulos a con halos periféricos de baja densidad; estos son heterogéneos y algunos contienen enzimas lisosómicas mientras otros son ricos en b tromboglobulina, factor plaquetario 4 (FP-4), fibrinógeno, factor de Von Willebrand, FVa, trombospondina, factor mitogénico y otras proteínas de la coagulación. La ausencia de estos gránulos provoca el interesante síndrome conocido como síndrome de la plaqueta gris.
EL SISTEMA TUBULAR DENSO
Este sistema funciona como un reservorio de calcio que es liberado al citoplasma por diferentes estímulos como el inositol trisfosfatado y el tromboxano A2, tras la estimulación de fosfolipasas.
SISTEMA DE CANALÍCULOS MEMBRANOSOS
Está conformado por una serie de canales interconectados y abiertos al exterior por diminutos poros de unos 25 nm aproximadamente que le confieren a la plaqueta su aspecto esponjoso y la capacitan en la incorporación de diversas moléculas que son absorbidas del plasma. Este sistema de vías permite la salida de los gránulos durante el proceso de secreción, aunque en rigor, en la actualidad no se han demostrado zonas de comunicación entre este sistema y las membranas de los gránulos.
Cuando se produce alteración del endotelio se establecen una serie de acontecimientos: el primero de ellos es la adhesión plaquetaria al subendotelio, interacción capaz de desencadenar la activación-agregación plaquetaria que predispone al proceso de secreción.
Las plaquetas son muy sensibles a diferentes estímulos y frente a una lesión endotelial reaccionan rápidamente, adhiriéndose al subendotelio; aquí participan receptores a nivel de membrana de las plaquetas y una serie de sustancias llamadas genéricamente adhesinas.
Los receptores plaquetarios están constituidos por las glicoproteínas llamadas integrinas y son los sitios de unión de las proteínas adhesivas. Este mecanismo también constituye el primer paso de la activación plaquetaria.
El intento más válido en la prevención de la trombosis arterial sería inhibiendo la adhesión plaquetaria.
Muchas de las funciones de las plaquetas (adhesión, agregación, asociación con otros elementos celulares) se realizan principalmente por la actividad funcional de las integrinas, las cuales favorecen la unión de las plaquetas al endotelio vascular (a través de las proteínas adhesivas).
Anomalías en el comportamiento de las integrinas pueden provocar el importante síndrome de plaquetas gigantes de Bernard-Soulier y la enfermedad de Glanzman, donde la unión plaqueta -fibrinógeno se encuentra severamente alterada; su característica principal es la deficiente retracción del coágulo.
Tanto el síndrome de Bernard-Soulier como la enfermedad de Glanzman se caracterizan por un tiempo de sangramiento prolongado.
Las integrinas están moduladas por concentraciones milimolares de calcio. En la actualidad se están realizando denodados esfuerzos en la caracterización de nuevos receptores adhesivos plaquetarios relacionados con las integrinas, puesto que esta nueva información facilitará la incorporación de medidas terapéuticas específicas en la enfermedad vascular.
MECANISMO DE ACTIVACIÓN PLAQUETARIA Y LIBERACIÓN DE GRÁNULOS
Un enorme número de sustancias pueden actuar sobre la plaqueta, propiciar su activación y su agregación, pero todas al parecer lo hacen activando fosfolipasas de la membrana celular que promueven la liberación de Ca+++, el cual por sí solo produce agregación y secreción (figura).
La activación plaquetaria se inicia por la unión de un agonista a la superficie plaquetaria (figura). La activación de las fosfolipasas C y A2 en las plaquetas se observa cuando análogos GTP no hidrolizables se introducen en las plaquetas permeabilizadas. Esto sugiere que las fosfolipasas C y A2 son reguladas por proteína G o de unión. Actualmente no se dispone de evidencias precisas respecto a qué proteínas regulan la activación de fosfolipasas C y A2 en las plaquetas.
Hasta el presente, 9 diferentes componentes de esta familia, conocida como proteína G han sido identificadas en las plaquetas y dependiendo de cada proteína G se puede estimular o inhibir el efecto.
Tras la activación por algunos agonistas, la fosfolipasa C actúa sobre el fosfatidil inositol 4,5 difosfato. La degradación del fosfoinosítido genera el 1,2 diacilglicerol (DG) y el inositol 1,4,5 trifosfato, (IP3).
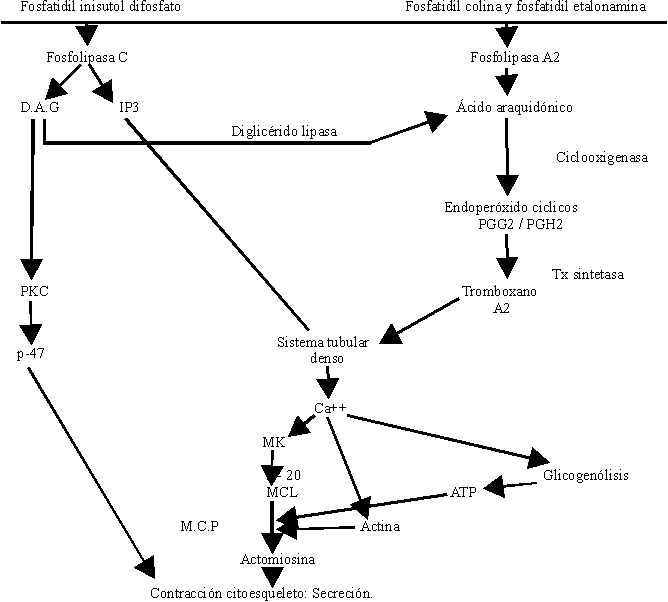
El DG activa a la proteína quinasa C (PKC) requiriendo dicha activación la presencia de calcio y fosfolípidos (fosfatidilserina y etanolamina). La PKC activa a una proteína de 47 kilodaltons, la cual es de gran importancia en el proceso de contracción del citoesqueleto plaquetario.
El IP3 se une a receptores específicos en el sistema tubular denso y libera al citosol hasta el 40 % del calcio almacenado. A su vez el DG es degradado por la diglicérido lipasa y se genera el 1-monoacilglicerol y ácido araquidónico (AA), pero esta no constituye la única fuente de AA en la plaqueta, también la activación de la fosfolipasa A2 promueve la liberación de AA de los fosfolípidos de membrana (fosfatidilcolina y fosfatidiletanolamina).
El AA es metabolizado a través de 2 mecanismos oxidativos: el de la ciclooxigenasa y de la lipooxigenasa. La vía de la ciclooxigenasa da lugar a la formación, primeramente, de los endoperóxidos cíclicos PGG2/PGH2 productos inestables y de capacidad agregante al actuar sobre receptores plaquetarios del tromboxano. Se metabolizan hacia productos estables finales las prostaglandinas: PGD2, PGE2 y PGF2 a y a productos intermedios inestables. Por efecto de la tromboxano sintetasa se forma un potente agente agregante plaquetario, vasoconstrictor, broncoconstrictor y que también aumenta la permeabilidad de las membranas, cuya vida media es de aproximadamente 30 segundos: el tromboxano (Tx A2).
La plaqueta no tiene equipo enzimático capaz de producir prostaciclina, por lo que constituye una estructura con capacidad exclusivamente agregante. El Tx A2 también promueve la liberación de calcio por el sistema tubular denso.
La activación de los mecanismos enzimáticos antes descritos promueve la liberación de calcio por el sistema de túbulos denso hacia el citosol plaquetario.
Las plaquetas en estado de reposo disponen en el citosol de 0,09-0,1 mM de calcio y después de la activación, en dependencia del agonista, se alcanzan valores de 0,2 mM con activadores débiles como serotonina y hasta incluso 3 mM con trombina. Este incremento se debe principalmente a la entrada de calcio extracelular tras la apertura de los correspondientes canales celulares.
El calcio procedente del líquido extracelular junto al liberado del retículo endoplásmico al citosol actúa como mensajero promoviendo señales eléctricas que inician la secuencia de las acciones trombocíticas:
- Cambio de forma. La plaqueta activada conlleva a un cambio morfológico, es decir, se esteriliza y emite pseudópodos, los cuales facilitan la interacción con otras plaquetas.
- Agregación plaquetaria. El complejo de GPIIb/IIIa cambia de forma dejando al descubierto el lugar de fijación que interacciona con proteínas adhesivas, en especial el fibrinógeno que se encuentran en la atmósfera periplaquetaria propiciando puentes de unión interplaquetarios, lo que da lugar al fenómeno físico de la agregación plaquetaria. El calcio sostiene a estos receptores unidos y es necesario en la unión del fibrinógeno al receptor. Las plaquetas se van uniendo unas a otras utilizando la molécula de fibrinógeno como cemento intercelular al unirse por el receptor plaquetario, la glicoproteína IIb-IIIa. La trombospondina está involucrada con la unión del fibrinógeno al complejo glicoproteico.
- Contracción del citoesqueleto. El Ca++ promueve la activación de la miosina quinasa y fosforila la cadena ligera de la miosina, de 20 kilodaltons de PM, a su vez se activan por acción del calcio las enzimas de la vía glucolítica promoviendo la formación de ATP, importante en la biosíntesis y en la contracción; el Ca++ disocia aquellas proteínas que cubren los extremos de la actina impidiendo su polimerización y el resultado es un incremento de actina filamentosa. La fosforilación de las cadenas ligeras de 20 kilodaltons de la miosina hace que los monómeros de miosina polimericen y adquieran una alta afinidad por la actina formándose la nueva molécula de actomiosina que provoca la contracción del citoesqueleto plaquetario. El Ca++ interviene en la activación de diversas enzimas que actúan sobre la materia filamentosa y retractil de la plaqueta.
- Secreción. Los filamentos retraídos centran los gránulos citoplasmáticos y al contactar las membranas de los gránulos con las invaginaciones de la membrana éstas se fusionan y posteriormente se lisan vertiéndose el contenido granular al exterior. Este fenómeno no bien conocido en lo que respecta a su mecanismo químico, se denomina exocitosis.
Conviene destacar que la adhesión plaquetaria, agregación y secreción actúan de manera sinérgica y casi simultáneamente promoviendo la formación del tapón plaquetario en la denominada hemostasia primaria.
REFERENCIAS BIBLIOGRÁFICAS
- Scazziota A, Altman R. Mecanismo de la hemostasia normal. Rev Iberoam Tromb Hemost 1994;7:95-109.
- Altman R, Scazziota A, Rouvier J. El mecanismo de la trombosis. Rev Iberoam Tromb Hemost 1994;7:110-7.
- Borrasca A, López Fernández A. La Hemostasia. Generalidades. Su relación con otros sistemas defensivos. En: López Borrasca A, ed. Enciclopedia Iberoamericana de hematología. 1 ed. Salamanca: Ediciones Universidad de Salamanca, 1992;vol 3:3-17.
- Meyer D, Girma JP. Von Willebrand factor: structure and functions. Thromb Haemost 1993;70:99-104.
- Fibronectins and Vitromectin. [Editorial]. Lancet 1989;1:474-6.
- Martínez J. Integrinas del sistema endotelioplaquetario. En: López Borrasca A, ed. Enciclopedia Iberoamericana de hematología. 1 ed. Salamanca: Ediciones Universidad de Salamanca, 1992;vol 3: 58-67.
- Prentice CRM. Pathogenesis of Thrombosis. Haemostasia 1990;20(suppl.1):50-9.
- Navarro J, César J. Estructura y morfología plaquetaria. En: López Borrasca A, ed. Enciclopedia Iberoamericana de hematología. 1 ed. Salamanca: Ediciones Universidad de Salamanca 1992;vol 3:76-82.
- Polak B, Ponzol P, Kolodie L. Endothelial cell and thrombosis. Rev Iberoam Thromb Hemost 1990;3:95- 105.
- García V, Navarro J. Adhesión, agregación y liberación plaquetarias. En:López Borrasca A, ed. Enciclopedia Iberoamericana de hematología. 1 ed. Salamanca: Ediciones Universidad de Salamanca, 1992;vol 3:112-121.
- Ordinas A, Escolar G, Batisda E, Castillo R. Actualización del papel de los componentes vasculares, plasmáticos y celulares en la regulación de la interacción de las plaquetas con el subendotelio. Sangre 1990;35:425-32.
Dr. Andrés O. Pérez Ruíz. Instituto Superior de Ciencias Médicas de La Habana. Calle 146 No.3102 esquina a 31, reparto Cubanacán, municipio Playa, Ciudad de La Habana, Cuba. CP11600.