










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versão On-line ISSN 1561-3011
Rev Cubana Invest Bioméd v.23 n.1 supl.1 Ciudad de la Habana ene.-mar. 2004
Trabajos de revisión
Instituto de Ciencias Básicas y Preclínicas Victoria de Girón
Algunos aspectos genéticos, moleculares y clínicos del cáncer colorrectal hereditario no polipoideo
Dra. Silvia Gra Menéndez y Lic. Diana Cruz-Bustillo Clarens
Resumen
Se hizo una revisión con el objetivo de profundizar en el conocimiento del cáncer colorrectal hereditario no polipoideo; resaltando su evolución histórica, así como sus características genéticas, moleculares y clínicas que condicionan el manejo clínico adecuado de los pacientes con esta afección y de los familiares en riesgo. La primera referencia al carácter hereditario del cáncer colorrectal fue a finales del siglo pasado, y el conocimiento actual de su base molecular, identificación clínica y diagnóstico genético es fruto de las investigaciones que se han llevado a cabo en este campo desde entonces. El cáncer colorrectal hereditario no polipoideo es una patología que representa aproximadamente 4 % del total de cáncer colorrectal.
DeCS: NEOPLASMAS COLORRECTALES NO POLIPOSIS HEREDITARIOS/ genética; NEOPLASMAS COLORRECTALES NO POLIPOSIS HEREDITARIOS/ patología; MUTACION; FACTORES DE RIESGO; GENES SUPRESORES.
El cáncer colorrectal (CCR) causa una significativa morbilidad y mortalidad al nivel mundial. En cerca de 80 % de los pacientes la enfermedad se presenta de forma esporádica, mientras que en 20 % restante existen componentes genéticos. El síndrome de CCR hereditario se subdivide en polipoideo, en el cual se incluyen la poliposis adenomatosa familiar (FAP), la poliposis familiar juvenil y el síndrome de Peutz-Jeghers y no polipoideo en el que se encuentra el cáncer colorrectal hereditario no polipoideo (HNPCC) o síndrome de Lynch.1 Además existen variantes de estas entidades que comparten la misma etiología genética pero tienen características clínicas distintivas. La FAP representa aproximadamente 1 % de todos los casos de CCR. La poliposis familiar juvenil, el síndrome de Peutz-Jeghers y la enfermedad inflamatoria del intestino juntas contribuyen en otro 1 %, y se estima que el HNPCC representa de 3 a 5 % (rango 1-13 %) de todos los CCR, siendo la entidad más frecuente dentro del síndrome de CCR hereditario.2
El objetivo de esta revisión es profundizar en el conocimiento del HNPCC resaltando su evolución histórica, así como sus características genéticas, moleculares y clínicas que condicionan el manejo clínico adecuado de estos pacientes y de los familiares en riesgo.
Historia
La historia de la enfermedad data de 1895, cuando el doctor Aldred Warthin conoció que su costurera estaba deprimida porque tenía la certeza de que moriría de cáncer de colon, gástrico o de útero, como le ocurría a la mayoría de los miembros de su familia. De hecho ella murió joven como consecuencia de un carcinoma endometrial. El doctor Warthin estudió esta familia y publicó en 1913 un artículo titulado Herencia con referencia a carcinomas.3 En el año 1966, el doctor Henry T. Lynch y otros describieron 2 familias, bajo el apelativo de síndrome de cáncer familiar, con los mismos hallazgos clínicos. El interés de estas descripciones radicaba en la amplia distribución anatómica del cáncer, su transmisión multigeneracional, la existencia de varios cánceres primarios en un mismo individuo y la alta incidencia de carcinomas de colon y endometrio.4 No fue hasta el año 1986 cuando estudios epidemiológicos, realizados en Finlandia y los EE. UU., demostraron que la predisposición al CCR se heredaba de forma dominante en la mayoría de los casos.1 Algunos años después, una serie de estudios internacionales documentaron la existencia del síndrome de cáncer familiar en el mundo. Durante esta fase de reconocimiento internacional, comenzó a emplearse el término cáncer colorrectal hereditario no polipoideo.5
Bases moleculares
En 1993 diversos autores sugirieron un nuevo mecanismo de tumorogénesis responsable de la aparición de la enfermedad. Se encontró una forma poco usual de mutación que afectaba la casi totalidad de los tumores de pacientes con HNPCC, y se detectaba por la aparición en el tejido tumoral de cambios en la longitud de microsatélites (regiones muy cortas del ADN que se repiten en tándem y que no codifican). Estas mutaciones fueron descritas como mutaciones somáticas ubicuas (USM),6 inestabilidad de microsatélites (MIN),7 o errores replicativos (RERs)8 y dieron lugar al modelo del fenotipo mutador como base de esta patología. Posteriormente se dilucidó que el origen de la inestabilidad de microsatélites se debía a mutaciones en los genes que intervienen en la reparación del ADN por apareamiento erróneo de bases.9-11 En el hombre los genes que cumplen esta función son: hMSH2 (human Mut S homolog 2), hMLH1 (human Mut L homolog 1), hPMS1 y 2 (human post meiotic segregation 1 y 2), hMSH6 (human Mut S homolog 6) y hMSH3 (human Mut S homolog 3) (tabla).12,13
Tabla. Genes humanos de reparación de bases mal apareadas
| Genes humanos de reparación de bases mal apareadas | |||
| Gen | Localización | No de exones | % de mutaciones en CCHNP |
| hMSH2 | 2p16-p15 | 16 | 31-50 |
| hMLH1 | 3p21.3-23 | 19 | 30-33 |
| HPMS1 | 2q31-33 | ? | 2-15 |
| HPMS2 | 7p22 | 15 | 4-5 |
| HMSH6 | 2p15 | ? | 0 |
| HMSH3 | 5q11-13 | ? | ? |
Tomado de González-Aguilera JJ, Fernández-Peralta AM. Genética del cáncer colorrectal. Prog Diag Prenat 1999;11(4):215-26.
El sistema de reparación por apareamiento erróneo de bases mejor caracterizado es el de la bacteria E. coli.14 En eucariontes este sistema es mucho más complejo y no ha sido totalmente dilucidado. Este mecanismo de reparación se inicia cuando el complejo de proteínas MSH2/MSH6 ó MSH2/MSH3 reconoce las bases mal apareadas y se une a esta zona del ADN. Estos 2 complejos tienen diferente afinidad para unirse al sitio de lesión de acuerdo con la naturaleza del daño. Posteriormente reclutan al complejo Mut L formado por las proteínas MLH1/PMS2. A continuación se produce la escisión de hasta 1 kb de ADN en la hebra mutada y luego este fragmento es resintetizado y ligado, por lo que queda corregido el daño (fig.). Se conoce poco acerca de la función de PMS1, así como del mecanismo que permite discriminar la hebra que porta la mutación.15
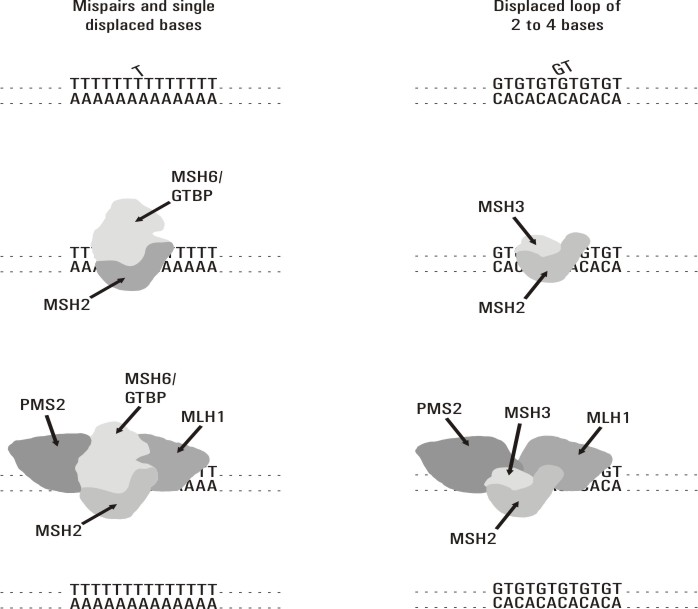
Fig. Mecanismo de reparación del DNA, por apareamiento erróneo de base, en humanos. Tomado de Wijnen Juul TH. Molecular Genetics of Hereditary Non-Polyposis Colorectal Cancer. [s.l]: [s.n]; [s.a]. 80p.
Aunque las mutaciones en los genes de reparación de apareamientos erróneos no son responsables directas de la iniciación y progresión del tumor, numerosos estudios indican que las células que presentan alteraciones en este mecanismo de reparación, acumulan mutaciones a un ritmo mucho mayor que las células normales. Además de las mutaciones en los microsatélites se producen alteraciones en secuencias codificadoras de genes supresores de tumores y protooncogenes, así como en genes que regulan el crecimiento y la muerte celular; por lo que se incrementa el potencial de transformación maligna de la célula afectada.16,17
Hay que señalar que en el HNPCC un alelo se encuentra inactivo debido a la mutación heredada, mientras que el segundo puede inactivarse debido a mutaciones somáticas, pérdida de heterocigosidad o hipermetilación.
Manifestaciones clínicas
El HNPCC se hereda de forma autosómica dominante y confiere 80 % de riesgo de desarrollar cáncer colorrectal a lo largo de la vida, comparado con 5 % de riesgo en la población general.18 La edad promedio en el momento del diagnóstico es de 44 años, 2 décadas antes que en los casos esporádicos.19 Los tumores predominan en la región proximal del colon (60-80 %), mientras que en los tumores esporádicos solo una pequeña parte (23-32 %) se localiza en el colon derecho. El riesgo de un CCR sincrónico en un paciente con HNPCC es 3 veces mayor que en los casos de CCR esporádicos (7,4 % para el gen hMLH1, 6,7 % para el gen hMSH2, y 2,4 % para los casos esporádicos). Mientras que el riesgo de desarrollar un CCR metacrónico es de 5-7 veces mayor que en los casos esporádicos.20 Histopatológicamente los CCR son poco diferenciados, abundantes en mucina extracelular y se distinguen por una respuesta linfocítica del huésped al tumor.21 Otros tipos de neoplasias son comunes en esta patología, y las localizaciones más frecuentes así como el riesgo de desarrollarlas a lo largo de la vida son: endometrio (39-60 %), ovario (9 %), estómago (12-19 %), sistema biliar (2-18 %), tracto urinario (4-10 %) e intestino delgado (1-4 %).22 La presencia o ausencia de tumores extracolónicos fue la base inicial para subdividir la enfermedad en síndrome de Lynch tipo I (solo CCR) y síndrome de Lynch tipo II (CCR y tumores extracolónicos); aunque actualmente ambas formas se expresan sencillamente como HNPCC o síndrome de Lynch. El riesgo de desarrollar neoplasias extracolónicas es mayor en los portadores de mutaciones en el gen hMSH2, que en el gen hMLH1 (48 vs. 11 %).4
El término no polipoideo en la enfermedad no es del todo correcto, porque se observan adenomas colorrectales con la misma frecuencia que en la población general. Además, como en los casos de CCR esporádicos, la transformación maligna se inicia a partir de un pólipo adenomatoso. Sin embargo, en pacientes con HNPCC estos pólipos aparecen a edades más jóvenes, tienen un comportamiento más agresivo y sufren transformación maligna con más frecuencia que los adenomas que se observan en la población general.1
Existen variantes de la enfermedad que se denominan síndrome de Muir-Torre y síndrome de Turcot. El síndrome de Muir-Torre es causado por los mismos defectos genéticos que se observan en el HNPCC, y presenta (además del espectro tumoral de la enfermedad) neoplasias sebáceas (incluidos adenomas, epiteliomas, carcinomas, queratoacantomas y quistes epidérmicos). No se conocen las causas de esta variedad fenotípica.18 Como síndrome de Turcot se conocen 2 entidades genéticas, una causada por mutaciones en el gen APC y la otra por mutaciones en los genes de reparación de apareamiento erróneo de bases; ambas presentan adenomas profusos en el colon y cánceres del SNC. Con el HNPCC se relaciona la segunda, y se caracteriza por la presencia de glioblastoma multiforme que aparece entre los 20 y 40 años, el glioblastoma multiforme que aparece en los casos esporádicos generalmente se desarrolla entre los 50 y los 80 años y tiene peor pronóstico que el relacionado con el HNPCC.18
Los pacientes con HNPCC tienen mejor pronóstico que aquellos con CCR esporádico. Se han realizado estudios comparativos de supervivencia en ambos grupos para los mismos estadios, y los pacientes con HNPCC tienen los porcentajes más altos.23 La explicación a este hecho todavía no ha podido ser totalmente dilucidada.
Diagnóstico
La ausencia de signos clínicos obvios dificulta la identificación y el control clínico de la enfermedad. La indicación más importante de un posible HNPCC proviene de una cuidadosa y extensa historia familiar. En 1991 el Grupo de Colaboración Internacional de HNPCC estableció los Criterios de Amsterdam para el diagnóstico clínico de esta patología.24 Deben encontrarse presentes todos los criterios siguientes:
- Al menos 3 familiares afectados con CCR, verificado histológicamente, uno debe ser familiar de primera consanguinidad de los otros 2 y se debe excluir la FAP.
- Al menos 2 generaciones sucesivas afectadas con CCR.
- Uno de los CCR o más debe diagnosticarse antes de los 50 años de edad del individuo.
En respuesta a que estos criterios son muy rigurosos, particularmente para aplicaciones clínicas y nuevas investigaciones, se han propuesto los Criterios de Amsterdam II.24 Estos incluyen los tumores extracolónicos del tipo comúnmente observado en el HNPCC, como son: cáncer de endometrio, tracto gastrointestinal superior y tracto urinario. En 1997 se establecieron los criterios de Bethesda que incluyen varias características clínico patológicas de la enfermedad; el paciente debe cumplir cualquiera de los siguientes:25
- Individuos con cáncer en familias que cumplen los criterios de Amsterdam.
- Individuos con 2 cánceres asociados a HNPCC, incluidos CCR sincrónicos y metacrónicos.
- Individuos con CCR y un familiar de primer grado con CCR y/o cáncer extracolónico asociado a HNPCC y/o un adenoma colorrectal, uno de los cánceres diagnosticado antes de los 45 años y el adenoma diagnosticado antes de los 40 años.
- Individuos con CCR o cáncer de endometrio diagnosticado antes de los 45 años.
- Individuos con CCR localizado en el lado derecho, con patrón no diferenciado, diagnosticado antes de los 45 años.
- Individuos con CCR del tipo célula en estampilla de sello de anillo, diagnosticado antes de los 45 años.
- Individuos con adenomas colorrectales diagnosticados antes de los 40 años.
Los criterios de Bethesda han sido modificados cambiando la edad del diagnóstico del adenocarcinoma de menos de 45 a menos de 50 años.25
Se recomienda que la prueba de inestabilidad en microsatélites deba ser el primer paso del estudio genético a realizar en pacientes sospechosos de padecer de HNPCC, porque más de 90 % de los CCR observados en la enfermedad la presentan. Se realiza en muestras de tumor de individuos afectados que cumplen los criterios de Amsterdam o los de Bethesda modificados. Si la presencia de inestabilidad en microsatélites es alta, entonces existen fuertes evidencias de que el paciente presente mutaciones germinales en los genes que intervienen en la reparación del ADN por apareamiento erróneo de bases; y se debe realizar el estudio molecular de los genes hMSH2 y hMLH1 (cuyas mutaciones son responsables de más de 95 % de los casos de CCHNP).1 El análisis comercial solo está disponible para la búsqueda de mutaciones en estos 2 genes. Si el individuo cumple cualquiera de los 3 primeros criterios de Bethesda modificados, entonces se puede realizar directamente el análisis molecular de los genes. Si no se dispone de tejido tumoral para realizar la prueba de inestabilidad en microsatélites, entonces el médico debe valorar la factibilidad de realizar directamente el estudio molecular. También se les debe realizar a familiares de primer grado (mayores de 20 años) de individuos que poseen una mutación conocida.2
Antes de realizar los estudios genéticos se debe obtener el consentimiento informado del paciente. Este debe incluir una descripción general y propósito de la prueba, descripción de la enfermedad, implicaciones de los resultados, sensibilidad y especificidad de la prueba, opciones para estimar el riesgo sin necesidad de la prueba y las acciones que el paciente podrá realizar si el resultado fuese positivo. Además se le debe brindar al paciente asesoramiento genético tanto antes como después de realizado el estudio genético, para así disminuir las reacciones psicológicas adversas que puedan presentarse.26-28
Seguimiento
Los estudios de detección precoz del CCR son muy importantes en esta patología. En ausencia de estudios genéticos los familiares de primer grado de individuos afectados tienen 50 % de riesgo de tener el gen mutado, por lo que se les recomienda la realización de una colonoscopia cada 1 ó 2 años, comenzando entre los 20 y 30 años, y anualmente después de los 40 años; o alternativamente cada 1 ó 2 años, comenzando a los 25 años.29 A los individuos portadores de la mutación se les debe realizar una colonoscopia anual, comenzando a los 25 años; ó 5 años antes de la edad que tenía en el momento del diagnóstico el paciente más joven de la familia.29
Los pacientes deben comprender que las estrategias de la colonoscopia son la extirpación de los pólipos, para prevenir la aparición del cáncer, y el diagnóstico precoz de los tumores; pero que una prevención completa es imposible de realizar.
Los estudios de detección precoz del cáncer endometrial deben realizarse anualmente, comenzando entre los 25 y 35 años. No existe un consenso sobre el método óptimo de pesquisaje, pero los más empleados son la aspiración endometrial y el ultrasonido transvaginal.30,31 Algunos expertos recomiendan realizar pruebas para detectar cánceres de ovario, estómago y genitourinario cuando estos tumores se han observado en la familia. Para detectar el tumor de ovario se realiza un examen ginecológico y ultrasonido transvaginal con la misma periodicidad que para el cáncer endometrial. En el caso del cáncer gástrico se realiza una gastroscopia cada 1-2 años, comenzando entre los 25 y 35 años. Para los tumores del tracto urinario y de células renales se realizan ultrasonidos y estudios de la orina cada 1 ó 2 años, comenzando entre los 30 y 35 años.30,31
Tratamiento
Se recomienda realizar una colectomía subtotal con anastomosis ileorrectal (y vigilancia posquirúrgica del recto cada 6 meses) cuando se desarrolla un CCR en el contexto de un HNPCC, debido a la alta incidencia de CCR metacrónicos observados en esta patología. Esta operación puede considerarse profiláctica en pacientes portadores de la mutación con adenomas diagnosticados durante el seguimiento, y constituye una opción para los portadores de la mutación sin evidencia de enfermedad al nivel de la mucosa.32,33
No existen suficientes evidencias para recomendar u oponerse a una histerectomía u ooforectomía como medida para reducir el riesgo de cáncer. Sin embargo, las mujeres portadoras de la mutación deben conocer que esta es una opción disponible.32,33
Conclusiones
Como se ha podido apreciar, el HNPCC es uno de los trastornos hereditarios causantes de cáncer más comunes en el hombre. En esta patología la identificación de familias en riesgo tiene gran trascendencia, debido a la alta frecuencia de aparición de cáncer en miembros asintomáticos. Para identificar las familias en riesgo el instrumento fundamental lo constituye la historia clínica familiar. Debido a la heterogeneidad de esta entidad, así como a la laboriosidad de su estudio clínico, patológico y genético es necesario que exista una colaboración entre diversos equipos multidisciplinarios; lo que permitirá un adecuado seguimiento y tratamiento de los individuos afectados.
Notables progresos se han realizado en la comprensión de las bases moleculares del HNPCC. Este progreso debe ser trasladado en los próximos años hacia nuevas técnicas de diagnóstico molecular, así como nuevas terapias farmacológicas que se traduzcan en un mayor beneficio para el paciente.
Summary
A review was made aimed at going deep into the knowledge about nonpolyposis hereditary cancer. Its historical evolution, as well as its genetic, molecular and clinical characteristics conditioning the clinical management of the patients with this affection and of the relatives at risk were stressed. The first reference to the hereditary character of colorectal cancer was made at the end of the last century. The current knowledge of its molecular base, clinical identification and genetic diagnosis are the result of the research carried out in this field since then. The nonpolyposis hereditary colorectal cancer is a pathology accounting for approximately 4 % of the total of colorectal cancer.
Subject headings: COLORECTAL NEOPLAMS, HEREDITARY NONPOLYPOSIS/ genetics; COLORECTAL NEOPLAMS, HEREDITARY NONPOLYPOSIS/ pathology; MUTATION; RISK FACTORS; GENES, SUPPRESSOR.Lynch HT, Smyrk T, Lynch J. An update of hereditary non-polyposis colorectal cancer (Lynch Syndrome). Cancer Genet Cytogenet 1997;93:84-99.
Recibido: 12 de octubre de 2003. Aprobado: 20 de noviembre de 2003.
Dra. Silvia Gra Menéndez. Calle 34 No. 2308 entre 23 y 25. municipio Playa, Ciudad de La Habana.

