










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión impresa ISSN 0864-0300
Rev Cubana Invest Bioméd vol.31 no.1 Ciudad de la Habana ene.-mar. 2012
ARTÍCULO ESPECIAL
Canalopatías iónicas: generalidades y panorámica actual
Ion channelopathies: Overview and current status
Dra. Margarita Dorantes Sánchez
Instituto de Cardiología y Cirugía Cardiovascular.
RESUMEN
Se presenta un panorama actual sobre las enfermedades de los canales iónicos cardíacos, con sus características comunes y algunas particularidades. Son síndromes arritmogénicos hereditarios por disfunciones en el tráfico de los canales iónicos al nivel de la membrana (mutaciones en los genes que codifican proteínas, con ganancia o pérdida de función), arritmias peligrosas para la vida, síncopes y muerte súbita, sin anormalidades estructurales gruesas detectables por los métodos convencionales. Estas enfermedades han experimentado un vertiginoso desarrollo en su entendimiento, diagnóstico y terapéutica en las últimas 2 décadas y se vislumbra un futuro promisorio con la genética molecular. Son diagnósticos de exclusión, se eliminan las cardiopatías estructurales, los trastornos electrolíticos y metabólicos, otras causas eléctricas y el empleo de fármacos antiarrítmicos. Se presentan en jóvenes aparentemente sanos, cuyo debut puede ser una arritmia ventricular maligna o un evento de muerte súbita, del cual solo es reanimado el 5 %. Comprenden un espectro clínico muy amplio, desde los asintomáticos (signos eléctricos, no síndromes), hasta los que fallecen. Su frecuencia real no se conoce debido a: muerte, diagnóstico erróneo, signos mínimos, intermitentes u ocultos. Se incorporan nuevas entidades, algunas se superponen y es muy difícil estratificar riesgo antes del debut. Se han creado registros internacionales. Se presentan los datos de nuestro Registro Nacional cubano de canalopatías en pacientes reanimados de eventos de muerte súbita seguidos durante 10 años.
Palabras clave: canalopatías iónicas, arritmias ventriculares malignas, muerte súbita.
ABSTRACT
An overview is presented of the current status of cardiac ion channel diseases, their common characteristics and some distinguishing features. Ion channelopathies are inherited arrhythmogenic syndromes caused by ion channel traffic dysfunctions at membrane level (mutations in protein-encoding genes with gain or loss of function), life-threatening arrhythmias, syncope and sudden death, without any gross structural abnormality detectable by conventional methods. The past two decades have witnessed speedy progress in the understanding, diagnosis and treatment of these diseases, a situation which will continue to be as promising in the future with the application of molecular genetics. They are exclusion diagnoses. Structural heart diseases, electrolyte and metabolic disorders, other electrical causes and the use of antiarrhythmic drugs are all discarded. They appear in seemingly healthy young persons, whose debut may be a malignant ventricular arrhythmia or a sudden death event, from which only 5 % are reanimated. They have a very broad clinical spectrum, ranging from asymptomatic cases (electrical signs, no syndromes) to fatal cases. Their actual frequency is unknown, due to: death, erroneous diagnosis, and minimal, intermittent or hidden signs. New diseases are incorporated, some overlap and it is very difficult to stratify risk before the debut. International registries have been developed. The paper presents the data contained in the Cuban National Channelopathy Register for patients reanimated from sudden death events and followed up for 10 years.
Key words: ion channelopathies, malignant ventricular arrhythmias, sudden death.
Actualidad de las canalopatías iónicas
El campo de las enfermedades de los canales iónicos ha tenido un impetuoso desarrollo en los últimos años y su futuro con la genética molecular es promisorio. Antes, solo algunos investigadores se interesaban en ellas; hoy, estos temas ocupan un lugar importante en todos los congresos de la especialidad cardiológica y en los de las subespecialidades.
Estas entidades presentan características comunes y al mismo tiempo, particularidades que las distinguen. Su verdadera frecuencia se desconoce porque algunos portadores fallecen sin tener diagnóstico; los signos eléctricos pueden ser intermitentes, transitorios, mínimos, ocultos, erróneamente interpretados o no vistos; los síncopes quedar sin explicación o darles una errónea; y algunos sujetos evolucionan sin síntomas.1-4
Aunque algunas de estas enfermedades ya se habían descrito, fue en 1995 cuando las canalopatías (CPs) se constituyeron ya como una nueva disciplina, y en el 2006 se les incluyó dentro de las cardiomiopatías primarias genéticas.5,6
Hoy, las publicaciones se multiplican, se incorporan nuevas entidades, se avanza en la comprensión de su fisiopatología, la genética y la presencia de alteraciones estructurales.7,8
Las publicaciones crecen de año en año, lo cual puede verse en las figuras 1 y 2, en cuanto al síndrome de Brugada (SBr) y al síndrome de QT largo (SQTL).


Lo normal
Cada latido cardíaco requiere una propagación precisa del potencial de acción a través del miocardio por la coordinación de 1 millón de canales iónicos en la superficie de cada cardiomiocito.
Las canalopatías
Algunos consideran que se trata de una enfermedad continuum entre diversas entidades, de variadas disfunciones en el tráfico de los canales iónicos (mutaciones con ganancia o pérdida de función), en distintas locaciones (epicardio, endocardio, ventrículo derecho o izquierdo, sistema especializado de conducción).7,8
A veces con una mixtura fenotípica de 2 entidades y puede suceder que diversas alteraciones lleven a un fenotipo común. Es decir, un mismo fenotipo puede deberse a alteraciones en distintos genes y una misma mutación dar lugar a variados fenotipos.
El SQTL es el paradigma de las CPs en varios aspectos: la primera estudiada (1957); el primer Registro Internacional (1979), cuyos primeros datos se publicaron en 1985 y que después sirvió de modelo para posteriores registros como el del síndrome de QT corto (SQTC), el del SBr, el de la fibrilación ventricular idiopática (FVI); la primera prueba genética (1991). Han existido grandes estudiosos a lo largo del tiempo: Jervell, Andersen, Lange-Nielsen, Timothy, Romano, Ward, Moss, Zareba, Schwartz, Keating, Towbin, Priori, Napolitano.9-12
De esa primera CP hasta una de las más recientemente aceptadas,

Las CPs son diagnósticos de exclusión; habrá que descartar alteraciones cardíacas estructurales, trastornos electrolíticos o metabólicos, otras causas eléctricas y el empleo de fármacos antiarrítmicos.
Alteraciones estructurales
Los conceptos han evolucionado con rapidez. Por ejemplo, su inclusión en la nueva clasificación de miocardiopatías en el 2006, donde se considera que constituyen una disfunción eléctrica aunque no mecánica, que son miopatías eléctricas e incluso a veces se acompañan de alteraciones estructurales miocárdicas no detectables por los métodos convencionales. No se sabe si lo estructural lleva a
Las arritmias son signo per se de disfunción eléctrica, reflejan enfermedad eléctrica miocárdica básica con o sin anormalidades estructurales. La enfermedad eléctrica primaria puede definirse como aquella con arritmias peligrosas para la vida, sin anormalidades gruesas ni histopatológicas. Son anomalías miocárdicas funcionales-estructurales al nivel molecular en la membrana, no identificables por métodos convencionales, biopsia ni necropsia.1-3,7,8
Definición
Se trata de condiciones cardíacas no estructurales, arrítmicas, hereditarias, por mutaciones de genes y desórdenes en los canales iónicos cardíacos con un miocito anormal y el corazón intacto en apariencia. Incluyen el SQTL, el SQTC, el SBr, la taquicardia ventricular polimórfica catecolaminérgica (TVPC), la enfermedad de Lev-Lenegre, el síndrome de muerte súbita (MS) nocturna inexplicada, la asociación de SBr y SQTC.1-3,7,8
Características comunes
Tienen puntos en común como: sujetos jóvenes por lo general; debut de arritmia ventricular maligna (AVM); pluralidad de causas; semejanzas en perfiles clínicos y AVM; iguales disparadores en algunas de estas; subregistro (por muerte, asintomáticos, signos mínimos, intermitentes, ocultos o que se escapan); fenotipos y genotipos complejos y heterogéneos; necropsia negativa; tendencia a recidivas y tormenta eléctrica; algunas AVM autolimitadas; síncope o paro cardíaco; reanimación exitosa en el menor número de sujetos; estudios invasivos y no invasivos de bajo valor predictivo; difícil estratificación de riesgo; signos premonitorios de compleja interpretación; distinción entre síndrome y signo; complejidad fisiopatológica; opciones terapéuticas a veces comunes, cardioversor-desfibrilador automático implantable (CDAI), ablación, quinidina, isoproterenol; confusiones diagnósticas con síncope vasovagal, infarto miocárdico, epilepsia, etc.1-3,7,8
Estructura
Cursan sin alteraciones estructurales detectables por métodos convencionales.
Desde 1991 se intentó cambiar el concepto del SQTL, en cuanto a que existían alteraciones estructurales. Solo 18 años después se reconocieron trastornos de la contracción y dispersión de la mecánica ventricular como integrantes del síndrome (no se conoce la contribución pronóstica de estos hallazgos). Se señaló que la enfermedad eléctrica primaria no es forzosamente una enfermedad eléctrica pura, sin disfunción mecánica. En el 2009 se afirmó que el SQTL a purely electrical disease? No anymore. Rosenbaum en 2010 se preguntó: Is long QT syndrome a disease of abnormal mechanical contraction?15,16
El SQTL presenta una contracción ventricular regional disincrónica, prolongada; aumento del índice de inhomogeneidad de los tiempos de contracción transmural y regional; dispersión del tiempo de contracción mecánica (incluso como mejor predictor que el QT largo); con diferencias entre los sintomáticos y los asintomáticos. La marcada prolongación del QT es signo pronóstico pobre, dicen algunos, y puede haber genética de QT largo con QT normal y SQTL con mutaciones no detectables.15,16
En general, la existencia de CP no implica que el corazón esté absolutamente libre de anormalidades estructurales finas (no como la causa directa de las AVM).13-18 En un estudio de SBr y resonancia magnética se encontraron alteraciones morfológicas-funcionales significativas de los ventrículos derecho e izquierdo. Los potenciales tardíos son relevantes en estos casos y ellos reflejan problemas de conducción.17
Los pacientes con signo eléctrico de Brugada, tienen un espectro clínico con diversa presentación. Algunos consideran que no es una enfermedad eléctrica pura. Frustaci y otros18 señalan que el patrón de supradesnivel del ST en precordiales derechas no es marcador de un síndrome específico, sino expresión eléctrica común de anormalidades estructurales en el ventrículo derecho, de origen genético, inflamatorio o infeccioso. Con lo cual variará el diagnóstico, la estratificación de riesgo y el proceder terapéutico.
Vínculos y solapamientos
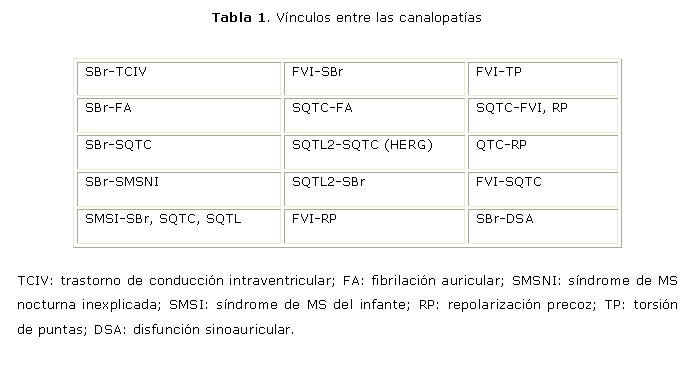
Entre las CPs existen vínculos, solapamientos, superposición y entrelazamientos. Algunos se resumen en la tabla 1.
Las CPs comparten gran variabilidad en su presentación clínica (fenotipos) y heterogeneidad genética, manifestaciones electrocardiográficas como fenotipos de variadas etiologías, variedad de enfermedades con la misma manifestación. A veces no deben verse como una enfermedad aislada.2,7,8,19-21
Como otro acercamiento entre ellas, podemos señalar las imágenes en espejo (Fig. 4).

En estas entidades existe una verdadera cascada de eventos: las mutaciones de los canales iónicos llevan a la alteración de las propiedades biofísicas y de la estructura de las proteínas y esto a una anormalidad estructural de los canales iónicos y de su arquitectura.2,7,8
Nuevas entidades que se añaden a las clásicas
Por otra parte, crece la lista de estos desórdenes arritmogénicos, hereditarios, poco comunes, congénitos, en plena expansión, donde se incluyen: síndrome de MS nocturna inexplicada, síndrome de MS del infante, FVI, FVI más RP. Y algunos casos de fibrilación y flutter auriculares, síndrome de preexcitación ventricular, DSA, paro auricular, bradicardia sinusal, taquicardia ventricular del tracto de salida del ventrículo izquierdo, bloqueo auriculoventricular. Entonces, las CPs son más frecuentes de lo que se pensaba.2-4,19,22-27
Como características generales puede señalarse que se trata de mutaciones en genes que codifican proteínas para el tránsito en los canales iónicos (Na+ K+, Ca++) en la membrana celular, que pueden llevar a enfermedad estructural y dispararse taquiarritmias ventriculares letales. Estos síndromes arritmogénicos hereditarios son miopatías eléctricas de origen genético, con riesgo de MS.1-4,6-8
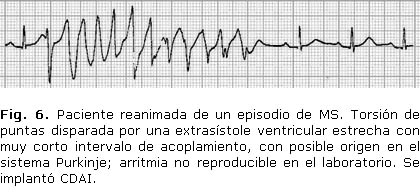
Las extrasístoles ventriculares de Purkinje suelen ser estrechas, con intervalo corto de acoplamiento. Desempeñan un papel en el inicio y el mantenimiento de las AVM, con capacidad automática, reentrante o disparada, y son malamente reproducibles en el laboratorio. Se invocan en
Estratificación de riesgo en estos sujetos
Lo más difícil en estas entidades es estratificar y modificar riesgo, en cuanto a episodios de AVM, recidivas y MS se refiere. Resulta fácil ir del sujeto reanimado a buscar signos eléctricos premonitorios aparecidos antes o después de la solución terapéutica. Pero lo contrario, ir de los signos premonitorios a predecir los candidatos a estos eventos malignos, es muy difícil y muchas veces imposible. En los pacientes de alto riesgo porque ya han tenido un primer episodio, es fácil establecer el pronóstico y la terapéutica, pero dentro de los de bajo riesgo, lo verdaderamente conflictivo resulta en quiénes van a presentarse los eventos malignos por primera vez o las recidivas. Se debe recordar que por lo demás, suelen ser sujetos absolutamente sanos.32-35
Utilidad de la estimulación eléctrica programada
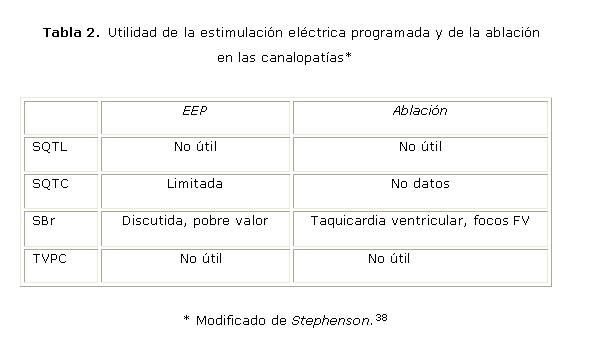
En cuanto a la estimulación eléctrica programada (EEP) para estratificar riesgo en las CPs, en general se considera que es poco o nada útil en los síndromes arritmogénicos hereditarios, con un papel incierto y limitado. Puede no inducirse la arritmia clínica o provocarse una no clínica. Será útil en casos seleccionados, por ejemplo, en arritmias supraventriculares asociadas. Los trabajos de los Brugada sí le conceden valor.36-38
En la tabla 2 se resume la utilidad de
Portadores asintomáticos
En cuanto al SBr asintomático, se ha señalado que el acercamiento terapéutico actual probablemente cause más daño que beneficio. Esto podría aplicarse también a lo que sucedería con los portadores asintomáticos de QT largo, QT corto, repolarización precoz.22,23,32-34
Otros conflictos
Algunos signos eléctricos pueden ser inocentes o culpables, ¿cómo diferenciar lo normal de lo arritmogénico? Por ejemplo, en cuanto al punto J, la memoria eléctrica, el intervalo QT corto o largo, las alteraciones de la onda T, las melladuras de
La onda J: ¿inocente, perversa?
Es la historia de 2 ondas J, opuestas en su trascendencia y riesgo. ¿Qué se le estudia?: configuración, extensión, amplitud, localización, evolución, asociación con intervalo QT corto. ¿Qué es?: signo, síndrome, marcador de malignidad, subpoblación de MS, hallazgo coexistente con otras entidades.23-27
Los debates vienen desde Shipley (1936) y Tomaszewski (1938), hasta hoy con los trabajos de Surawicz y de Antzelevitch (2011). El SBr representaría una cuarta variante de repolarización precoz, limitada a las derivaciones precordiales derechas.24,25
Papel de la quinidina
Es el fármaco antiarrítmico más antiguamente conocido, el más efectivo y a veces el único a emplear en CPs tales como los síndromes de Brugada, de repolarización precoz, de QT corto y
Conducta terapéutica
Si algunos SBr son de causa inflamatoria o infecciosa, no todos requerirían CDAI. Tampoco todos los SQTL, sin embargo, aumenta el número de los dispositivos implantados, sin recordar sus frecuentes complicaciones, sobre todo a edades jóvenes. De tal manera que existe un European LQTS-CDAI Registry, para apreciar cuándo deben implantarse. La terapia apropiada se predice por: menos de 20 años de edad en el primoimplante, intervalo QTc mayor de 500 ms, paro cardíaco previo y eventos cardíacos a pesar de las terapias. Se considera que a los 7 años, no hay choques apropiados en ningún paciente sin alguno de estos factores y los apropiados llegan al 70 % en aquellos que tienen todos los factores.12
En el European LQTS- CDAI Registry se discute cómo están estos pacientes y qué pasa con ellos (233 sujetos, del 2002 al 2010): el 9 % asintomático cuando se implantó el dispositivo, ¿eran de alto riesgo?, ¿se consideró el pro y el contra? Los choques fueron apropiados en el 28 % y hubo eventos adversos en el 25 %.12
En un paciente asintomático, algunas enfermedades identificadas por pruebas genéticas pueden ser subclínicas y nunca dar síntomas; en ellos el inicio del tratamiento puede llevar a mayor riesgo que beneficio.7,8
En el SQTL debe implantarse el dispositivo en todos los casos con otro tratamiento y paro cardíaco, en muchos sobrevivientes de paro cardíaco sin tratamiento (excepto si existe una causa evitable), síncope con beta bloqueadores, 2 mutaciones-síncope-beta bloqueadores, sujetos asintomáticos-QT mayor de 550 ms, inestabilidad eléctrica, T alternante, pausas largas.12
Nuestros datos
El Instituto de Cardiología y Cirugía Cardiovascular es centro nacional de referencia de sujetos reanimados de episodios de MS arrítmica, sin cardiopatía estructural demostrable por métodos convencionales. El Registro Nacional Cubano de estos pacientes incluye 106 pacientes, 92 corresponden a CPs (60 del sexo maculino y 32 del femenino). No se incluyen los no reanimados, los asintomáticos, los que no presentaron episodio de MS, los no diagnosticados. Por lo tanto, el número real es mayor del apreciado, no se conoce.
En el grupo de las CPs, tuvieron recidivas de AVM 77,2 %, tormenta eléctrica 8,7 % e inducibilidad de la arritmia en
Correspondieron a FVI 37 pacientes, SBr 31, SQTL 19, taquicardia ventricular idiopática 3, SQTC 1, TVP 1. Las arritmias presentes fueron: FV, torsión de puntas, taquicardia ventricular, TVP. Las soluciones terapéuticas: CDAI 76, fármacos antiarrítmicos 45, marcapaso 10, ablación 3. Un paciente pudo recibir más de una opción.
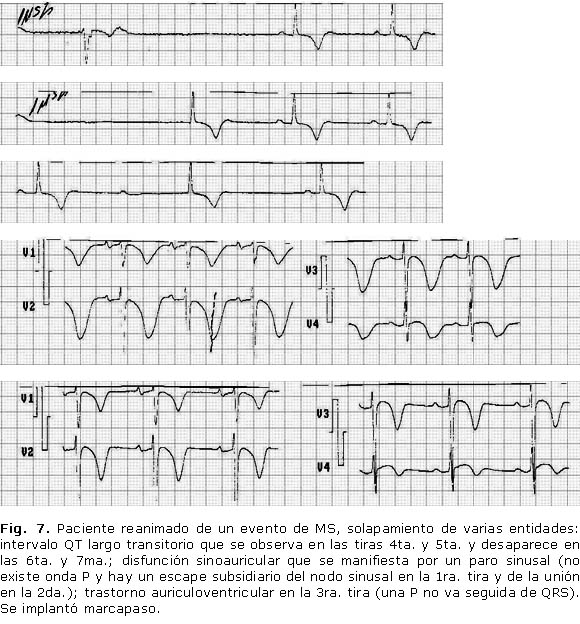
Se presentan trazados de algunos de los pacientes de nuestra serie (Figs. 5, 6 y 7).

Sinopsis
En cuanto a las CPs, mencionemos algunos puntos esenciales y otros conflictos: inclusión de nuevas entidades; sujetos asintomáticos que plantean problemas al precisar el riesgo y decidir la opción terapéutica; diferencia entre signo y síndrome; espectro amplio; AVM que pueden ser autolimitadas; importante papel del sistema Purkinje como disparador de las arritmias; poco valor predictivo de la estimulación eléctrica; frecuentes recidivas y tormenta eléctrica; vínculos y solapamientos entre ellas; necesidad de estudios multicéntricos y multipaíses para arribar a conclusiones en el menor tiempo; posible alteración estructural solo determinada por métodos muy finos y no convencionales; signos intermitentes, transitorios, mínimos y enmascarados, que dificultan el diagnóstico; muy difícil estratificación de riesgo en estos pacientes jóvenes por lo general y aparentemente sanos, en quienes el debut puede ser una AVM; dificultad para evaluar los signos premonitorios; opciones terapéuticas a veces compartidas (CDAI, quinidina, isoproterenol en el SBr, SQTC, FVI, repolarización precoz); selección cuidadosa del CDAI en el SQTL, no siempre necesario.
En general
Pueden ser diferentes caras de una misma moneda: genéticas, iónicas, celulares y plataforma arritmogénica.
Las CPs constituyen un espectro continuo de expresión fenotípica. Pocas pruebas son positivas de mutaciones: puede haber clínica sin mutaciones y mutaciones sin clínica. Priori11 ofrece una visión unitaria de los síndromes arritmogénicos hereditarios y describe los canales iónicos como complejos macromoleculares. Las proteínas intracelulares tienen un papel en su patogenia y una puede intervenir en más de una enfermedad. Así, no solo los canales iónicos, sino algunas proteínas interactuantes con ellos, estarían envueltas en estos síndromes. Puede decirse, al modo de Priori, que en ellos la electrofisiología cardíaca se alimenta de la electrofisiología molecular (revolución iniciada por Keating en 1991 en el SQTL), donde se unen clínicos, biólogos moleculares, genetistas y electrofisiólogos celulares.
REFERENCIAS BIBLIOGRÁFICAS
1. Consensus Statement of the Joint Steering Committees of the Unexplained Cardiac Arrest Registry of Europe of the Idiopathic Ventricular Fibrillation Registry of the United States: Survivors of out-of-hospital cardiac arrest with apparently normal heart. Need for definition and standardized clinical evaluation. Circulation. 1997;95:265-72.
2. Antzelevitch C. Síndrome de Brugada. Del laboratorio a la clínica. Eds. Asociados y directores de la edición española. Brugada P, Brugada J, Brugada R. Madrid: J & C. Ediciones Médicas SL. 2006. pp. 47, 48, 59, 69, 111, 119, 160.
3. Priori SG, Napolitano C, Grillo M. Concealed arrhythmogenic syndromes: the hidden substrate of idiopathic ventricular fibrillation. Cardiovasc Res. 2001;50:218-23.
4. Priori SG, Aliot E, Blomstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, et al. Task force on sudden cardiac death of the European Society of Cardiology. Eur Heart J. 2001;22:1374-450.
5. Corrado D, Basso C, Thiene G. Is it time to include ion channel diseases among cardiomyopathies? J Electrocardiol. 2005;38(4 suppl):81-7.
6. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation. 2006;113:1807-16.
7. Cerrone M, Priori SG. Genetics of sudden death: focus on inherited channelopathies. Eur Heart J. 2011;32:2109-18.
8. Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ Arrhythm Electrophysiol. 2009;2:6-15.
9. Moss AJ, Schwartz PJ, Crampton RS, Locati E, Carleen E. The long QT syndrome: a prospective international study. Circulation. 1985;71:17-21.
10. Moss AJ, Schwartz PJ. 25th Anniversary of the International Long-QT Syndrome Registry. An ongoing quest to uncover the secrets of long-QT syndrome. Circulation. 2005;111:1199-201.
11. Priori SG. The fifteen years of discoveries that shaped molecular electrophysiology. Time for appraisal. Circ Res 2010;107:451-6.
12. Schwartz PJ, Spazzolini C, Priori SG, Crotti L, Vicentini A, Landolina M, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them? Data from the European Long-QT Syndrome Implantable Cardioverter-Defibrillator (LQTS ICD Registry). Circulation. 2010;122:1272-82.
13. Hoogendijk MG, Opthof T, Postema PG, Wilde AA, de Bakker JM, Coronel R. The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ Arrhythm Electrophysiol. 2010;3:283-90.
14. Brugada P. Commentary on the Brugada ECG pattern. A marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ Arrhythm Electrophysiol. 2010;3:280-2.
15. De Ferrari GM, Schwartz PJ. Long QT syndrome, a purely electrical disease? Not anymore. Eur Heart J. 2009;30:253-5.
16. Rosenbaum DS. Is long QT syndrome a disease of abnormal mechanical contraction? Circulation. 2010;122:1353-4.
17 Papavassiliu T, Veltmann C, Doesch C, Haghi D, Germans T, Schoenberg SO, et al. Spontaneous type 1 electrocardiographic pattern is associated with cardiovascular magnetic resonance imaging changes in Brugada syndrome. Heart Rhythm. 2010;7:1790-6.
18. Frustaci A, Priori SG, Pieroni M, Chimenti C, Napolitano C, Rivolta I, et al. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation. 2005;112:3680-7.
19. Viskin S, Zeltser D, Ish-Shalom M, Katz A, Glikson M, Justo D, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm. 2004;1:587-91.
20. Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. JACC. 2011;57:802-12.
21. Viskin S. The QT interval: too long, too short or just right. Heart Rhythm. 2009;6:711-5.
22. Viswanathan MN, Page RL. Short QT: when does it matter. Circulation. 2007;116:686-8.
23. Stern S. Clinical aspects of the early repolarization syndrome: a 2011 update. Ann Noninvasive Electrocardiol. 2011;16:192-5.
24. Surawicz B, Macfarlane PW. Inappropriate and confusing electrocardiographic terms. J-wave syndromes and early repolarization. JACC. 2011;57:1584-6.
25. Antzelevitch C, Yan GX, Viskin S. Rationale for the use of the terms J-wave syndromes and early repolarization. JACC. 2011;57:1587-90.
26. Viskin S. Idiopathic ventricular fibrillation “le syndrome d'Haissaguerre” and the fear of J waves. JACC. 2009;53:620-2.
27. Benito B, Guasch E, Rivard L, Nattel S. Clinical and mechanistic issues in early repolarization. Of normal variants and lethal arrhythmia syndromes. JACC. 2010;56:1177-86.
28. Boyden PA, Hirose M, Dun W. Cardiac Purkinje cells. Heart Rhythm. 2010;7:127-35.
29. Ideker RE, Kong W, Pogwizd S. Purkinje fibers and arrhythmias. PACE. 2009;32:283-5.
30. Scheinman MM. Role of the His-Purkinje system in the genesis of cardiac arrhythmia. Heart Rhythm. 2009;6:1050-8.
31. Baher AA, Uy M, Xie F, Garfinkel A, Qu Z, Weiss JN. Bidirectional ventricular tachycardia: ping pong in the His-Purkinje system. Heart Rhythm. 2011;8:599-605.
32. Viskin S, Rogowski O. Asymptomatic Brugada syndrome: a cardiac ticking time-bomb? Europace. 2007;9:707-10.
33. Viskin S, Rosso R. Risk of sudden death in asymptomatic Brugada syndrome. Not as high as we thought and not as low as we wished... but the contrary. JACC. 2010;56:1585-8.
34. Viskin S. Brugada syndrome in children: don't ask, don't tell? Circulation. 2007;115:1970-2.
35. Surawicz B. Brugada syndrome: manifest, concealed, “asymptomatic”, suspected and simulated. JACC. 2001;38:775-7.
36. Paul M, Gerss J, Schulze-Bahr E, Wichter T, Vahlhaus C, Wilde AAM, et al. Role of programmed ventricular stimulation in patients with Brugada syndrome: a metanalysis of worldwide published data. Eur Heart J. 2007;28:2126-33.
37. Brugada P, Brugada R, Mont L, Rivero M, Geelen P, Brugada J. Natural history of Brugada syndrome: the prognostic value of programmed electrical stimulation of the heart. J Cardiovasc Electrophysiol. 2003;14:455-7.
38. Stephenson EA, Berul CI. Electrophysiological interventions for inherited arrhythmia syndromes. Circulation. 2007;116:1062-82.
39. Vardas PE, Kanoupakis EM. Old drugs never die; they just fade away. Heart Rhythm. 2010;7:864.
40. Viskin S, Belhassen B, Wilde AA. Irreplaceable antiarrhythmic medications are disappearing: the case of Quinidine. Heart Rhythm. 2010;7:863.
41. Viskin S, Antzelevitch C, Márquez MF, Belhassen B. Quinidine: a valuable medication joins the list of “endangered species”. Europace. 2007;9:1105-6.
42. Viskin S, Wilde AAM, Tan HL, Antzelevitch C, Shimizu W, Belhassen B. Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry. Heart Rhythm. 2009;6:401-4.
Recibido: 24 de febrero del 2012.
Aprobado: 1 de abril del 2012.
Dra. Margarita Dorantes Sánchez. Instituto de Cardiología y Cirugía Cardiovascular.

{kind=link}
{kind=link}