










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión impresa ISSN 0864-0300
Rev Cubana Invest Bioméd vol.31 no.2 Ciudad de la Habana abr.-jun. 2012
REVISIÓN
Taquicardia ventricular polimórfica catecolaminérgica
Catecholaminergic polymorphic ventricular tachycardia
Dra. Marleny Cruz Cardentey
Departamento de Arritmias y Marcapasos. Hospital Clinicoquirúrgico “Hermanos Ameijeiras”.
RESUMEN
La taquicardia ventricular polimórfica catecolaminérgica es una canalopatía caracterizada por la inducción de arritmias ventriculares polimórficas en presencia de catecolaminas. Deberá sospecharse en todo paciente joven, en especial niño o adolescente, que presente síncopes relacionados con el ejercicio físico o el estrés emocional, que no tenga cardiopatía estructural y que su electrocardiograma muestre un intervalo QT normal. Es poco frecuente, pero importante por el riesgo elevado de muerte súbita, que en ocasiones puede ser el debut. Las arritmias ventriculares son polimórficas o bidireccionales, fácilmente inducibles con el ejercicio físico y con infusión de isuprel, tienen un umbral predecible y una complejidad progresiva. Los antecedentes patológicos familiares de muerte súbita se observan entre el 30 y 40 % de los pacientes. Se han identificado 2 mutaciones genéticas causantes de la entidad (receptores de rianodina 2, con herencia autosómica dominante y calsecuestrina 2, con herencia autosómica reseciva); pero solo entre 50-55 % de los enfermos se ha testado una mutación causal. Las mutaciones condicionan la fuga de Ca2+ del retículo sarcoplásmico que favorece el origen de posdespolarizaciones tardías, las que inducirán la actividad ectópica ventricular. Los â-bloqueadores son el tratamiento de elección. El desfibrilador automático implantable está indicado en los pacientes recuperados de un evento de muerte súbita y en los sintomáticos a pesar del tratamiento farmacológico. La denervación simpática cardíaca izquierda, el verapamilo, la flecainida y la propafenona, son opciones alternativas en los sintomáticos a pesar del uso de β-bloqueadores.
Palabras clave: taquicardia ventricular polimórfica catecolaminérgica, muerte súbita, canalopatías.
ABSTRACT
Catecholaminergic polymorphic ventricular tachycardia is a channelopathy characterized by the induction of polymorphic ventricular arrhythmias in the presence of catecholamines. It should be suspected in any young patient, especially a child or adolescent, presenting with syncope associated with physical exercise or emotional stress, with no structural heart disease and an ECG showing a normal QT interval. It is a rare disease, its importance lying in the high risk of sudden death, which may sometimes be its debut. Ventricular arrhythmias may be polymorphic or bidirectional. They are highly inducible by physical exercise and Isuprel infusion, their threshold is predictable and their complexity progressive. A family history of sudden death is reported in 30 to 40 % of patients. Two genetic mutations have been identified as causes of the condition (ryanodine receptor 2 with autosomal dominant inheritance and calsequestrin 2, with autosomal recessive inheritance). However, a causal mutation has been found in only 50-55 % of patients. Mutations influence sarcoplasmic reticulum Ca 2+ leak, facilitating the appearance of late post-depolarisations, which will in turn induce ventricular ectopic activity. Beta-blockers are the treatment of choice. The automatic implantable defibrillator is indicated in patients recovered from a sudden death event and in those who remain symptomatic despite medical therapy. Left cardiac sympathetic denervation, verapamil, flecainide and propafenone are alternative options for patients who remain symptomatic despite the use of beta-blockers.
Key words: catecholaminergic polymorphic ventricular tachycardia, sudden death, channelopathies.
INTRODUCCIÓN
La taquicardia ventricular polimórfica catecolaminérgica (TVPC) es una enfermedad eléctrica primaria del corazón, de carácter hereditario, y caracterizada por la inducción de arritmias ventriculares polimórficas en presencia de catecolaminas (ejercicio físico y estrés) y por la ausencia de cardiopatía estructural.1
En 1975 Reid y otros2 realizan la primera descripción de la entidad en una niña de 6 años; posteriormente en 1978, Coumel y otros3 reportan en 4 niños la inducción una taquicardia ventricular polimórfica promovida por catecolaminas. Desde entonces, se han publicado solo casos aislados y series con un número reducido de pacientes.4-6 Hasta la fecha, las series más amplias son las presentadas por Leenhardt y otros7 en 1995 y por Priori y otros8 en el 2002, con 21 y 30 pacientes, respectivamente.
Sin embargo, no fue hasta el año 2001 cuando Priori9 y Lahat10 describen, respectivamente, las mutaciones en el gen del receptor de rianodina tipo 2 (RyR2) y en el gen de la calsecuestrina cardíaca (CASQ2), involucradas en la patogénesis de
Clínica
Los pacientes con TVPC sufren cuadros sincopales recurrentes desencadenados por el ejercicio físico o tensiones emocionales, y en algunos casos la muerte súbita es su forma de presentación.
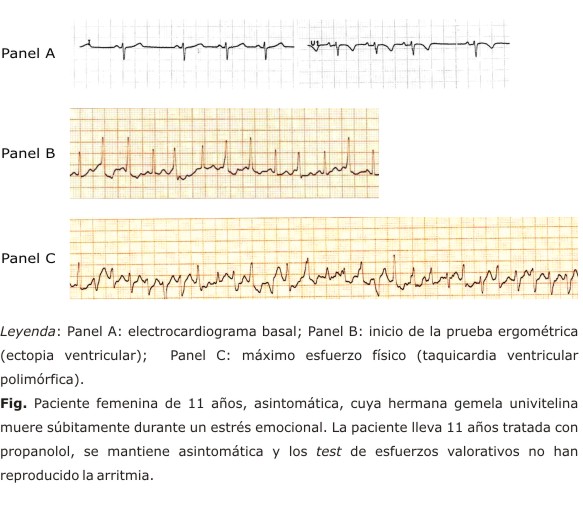
Las taquicardias ventriculares desencadenadas por el ejercicio o el estrés emocional tienen ciertas particularidades: son polimórficas y con frecuencia pueden ser bidireccionales, (alternancia del QRS en 180° de latido en latido) similar a la taquicardia observada durante la intoxicación digitálica.2,5 Resultan fácilmente inducible y reproducible con el ejercicio físico. Existe un umbral predecible de frecuencia cardíaca para el inicio de la secuencia arrítmica, el cual se encuentra entre 100-120 latidos por minuto de taquicardia sinusal. Presenta una sucesión característica, iniciada por extrasistolia ventricular aislada, y en la medida en que aumentan los niveles de catecolaminas, progresa la complejidad de la arritmia, hasta finalizar en taquicardia polimórfica y ocasionalmente en fibrilación ventricular (fig.) Durante el ejercicio también pueden desencadenarse arritmias supraventriculares.13
En el registro de Holter la bradicardia sinusal es un hallazgo reportado en varios pacientes. Sin embargo, el tratamiento con β-bloqueadores es por lo general bien tolerado, sin ocasionar bradicardia severa sintomática.7,14 En el electrocardiograma basal se han descrito ondas U prominentes y ondas T negativas de V1-V3, pero estos elementos son considerado normales en sujetos jóvenes.7,15 En sentido general, estas alteraciones eléctricas son poco frecuentes y no suficientemente específicas para un diagnóstico.
BASES GENÉTICAS Y ELECTROFISIOLÓGICAS
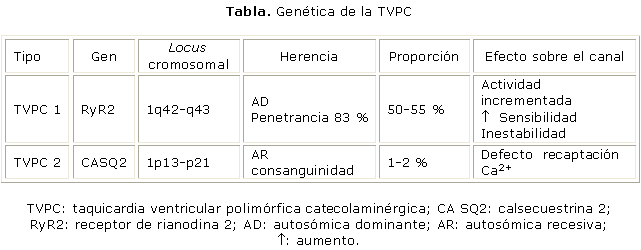
Los antecedentes patológicos familiares de muerte súbita existentes entre el 30 al 40 % de los pacientes con TVPC,8 hizo sospechar sobre una probable base genética en esta entidad (tabla).
La primera mutación testada se encuentra en el gen que codifica al RyR2.9 Se han caracterizado 130 mutaciones en el RyR2, que representan entre el 50-55 % del total de las identificadas en
El RyR2 es un elemento clave en el acoplamiento del proceso excitación-contracción. En la fase 2 del potencial de acción se activan los canales de Ca2+ tipo L y se produce la entrada de este ión al medio intracelular. Las concentraciones elevadas de Ca2+ activan a su vez a los RyR2 y se libera el ión almacenado en el retículo sarcoplásmico (liberación de Ca2+ inducida por Ca2+). En diástole, el Ca2+ es removido del medio intracelular por 3 proteínas en particular: bomba de Ca2+ del retículo sarcoplásmico o SERCA-2, intercambiador de Na+/Ca2+ y la bomba de Ca2+ de la membrana celular.
Las mutaciones descritas llevan a una actividad incrementada en reposo, a un aumento de la sensibilidad a determinados estímulos y a una inestabilidad del canal iónico, situaciones que generan una sobrecarga intracelular de Ca2+.16-19
El segundo gen asociado con
Se han testado 7 mutaciones en el CASQ2, las que representan menos del 5 % de las causantes de la enfermedad y condicionan
Se sospecha que otros genes puedan estar involucrados en la genética de
Hasta la fecha, la correlación genotipo-fenotipo en
Las mutaciones genéticas descritas en
La primera descripción de la taquicardia bidireccional como manifestación de la intoxicación digitálica fue en el año 1922; desde entonces el esclarecimiento de sus mecanismos electrofisiológicos ha suscitado el interés de los cardiólogos.27 Ha sido descrita en otros contextos clínicos: parálisis periódica familiar por hipopotasemia,28 síndrome de Andersen Tawil,29 miocarditis fulminante30 y TVPC.
Dentro de las teorías propuestas para explicar tan complejo fenómeno eléctrico se encuentran: arritmias supraventriculares con bloqueo alternante del fascículo anterior y posterior,31 único foco en el has de His o en sus ramas con bloqueo alternante de los fascículos izquierdos, único o doble foco ectópico en el sistema His-Purkinje distal.26
En contraste con estos complejos y no esclarecedores mecanismos, recientemente se ha publicado una nueva teoría electrofisiológica sobre el origen de la taquicardia bidireccional. Un simple mecanismo de ping-pong, en el cual las posdespolarizaciones tardías inducen la actividad gatillada, con diferentes umbrales de frecuencia cardíaca en las diferentes regiones del sistema His-Purfinke y del miocardio ventricular para dispararla. El bigeminismo reciprocante se establece entre 2 focos ectópicos que no precisan estar situados en ramas o ventrículos opuestos, por ejemplo: 2 focos gatillados localizados en el mismo ventrículo o en epicardio y endocardio.26,32 El patrón más común de taquicardia bidireccional en humanos es el que se presenta con bloqueo de rama derecha y con desviación axial derecha e izquierda alternante, como expresión de un bigeminismo reciprocante localizado en región distal del fascículo izquierdo anterior y posterior.
DIAGNÓSTICO
Para confirmar el diagnóstico se precisa reproducir la arritmia ventricular mediante una prueba ergométrica o una infusión de isoprotenerol. El test de Holter puede recoger actividad ventricular ectópica o taquicardia ventricular cuando el paciente se expone a alguna descarga adrenérgica. Dada la alta sensibilidad de la ergometría y la infusión de isoprotenerol (100 y 75 %, respectivamente) para reproducir las arritmias ventriculares en
El diagnóstico diferencial debe realizarse en especial con el síndrome de QT largo (SQTL), pues ambas miopatías eléctricas compartes ciertas características clínicas: edad de debut, ausencia de cardiopatía estructural, antecedentes familiares de síncope o muerte súbita, aparición de los síntomas en relación con el estrés físico y emocional y carácter polimórfico de las arritmias ventriculares. La principal diferencia entre ambas canalopatías es la existencia de un intervalo QT prolongado en el SQTL, aunque la existencia de un intervalo QT normal no excluye el diagnóstico de esta entidad (5-10 % de portadores genéticos en el SQTL muestran un intervalo QT normal).34
En
Debe diferenciarse además de otros tipos de taquicardias bidireccionales, como el síndrome de Andersen-Tawil, caracterizado por una prolongación del intervalo QT, ondas U prominentes, patrón dismórfico facial, parálisis periódica y taquicardia bidireccional más lenta y con intervalo de acoplamiento más largo.35 En los pacientes con mutaciones del RYR2 deberá evaluarse cuidadosamente la anatomía y la función del ventrículo derecho, con el fin de descartar una rara variante de displasia arritmogénica de ventrículo derecho.
Si la mutación ha sido testada en un individuo, a todos los familiares de primera línea se les deberá ofrecer un estudio genético-molecular, siempre que se disponga de un laboratorio especializado en el medio. Si se desconoce la mutación o no se dispone del test genético-molecular, los familiares serán evaluados con electrocardiograma basal, prueba de esfuerzo y monitorización de Holter.
TRATAMIENTO
Basados en los mecanismos fisiopatológicos de
Se recomienda la dosis máxima tolerada, la cual se reajustará con periodicidad, teniendo presente el incremento del peso corporal en los niños y adolescentes.37 La efectividad terapéutica será evaluada periódicamente (entre 6 y 12 meses) con prueba ergométrica y monitorización de Holter, valorando el tiempo entre la última dosis y el horario de realización de la ergometría. En pacientes bajo tratamiento con β-bloqueadores, el test será limitado por la máxima carga tolerada y no por la frecuencia máxima ajustada a la edad.38
Los deportes competitivos y los ejercicios físicos extremos están siempre contraindicados. Todos los pacientes con arritmias inducidas por el ejercicio deben limitar la actividad física, con la excepción de los que muestren una supresión de las arritmias con el tratamiento farmacológico.38
En los pacientes recuperados de un episodio de muerte súbita está indicado el implante de una cardio-desfibrilador para la prevención secundaria de eventos arrítmicos futuros. En los individuos que no toleren dosis altas de β-bloqueadores o que no exista un adecuado control de las arritmias con el tratamiento farmacológico, el implante de un cardio-desfibrilador se recomienda como prevención primaria de la muerte súbita arrítmica.8,33,39
No es despreciable el efecto pro-arrítmico de los desfibriladores en esta miopatía eléctrica. El miedo en los niños a las descargas eléctricas del dispositivo produce la liberación de catecolaminas, que generarán más eventos arrítmicos, instaurándose un círculo vicioso. (miedo-catecolamina-arritmia-descarga-miedo).
Una opción terapéutica alternativa es la denervación simpática cardíaca. Fundamentado por los mecanismos fisiopatológicos de
En 3 trabajos se ha reportado la efectividad parcial del verapamilo en los pacientes con TVPC, aunque estos reportes no han sido adecuadamente confirmados.33,41,42 Se desconocen además los efectos sobre la contractilidad miocárdica, del uso crónico de dosis altas de β-bloqueadores y anticálcicos.
Recientemente se demostró que la flecainida, mediante la inhibición de los canales del RyR2 liberadores de Ca+2, previene los eventos arrítmicos en modelos de ratón con TVPC y en humanos.43 Posteriormente Hwang y otros44 investigaron la efectividad de los fármacos antiarrítmicos del grupo I en esta canalopatía. Solo la flecainida y la propafenona produjeron un bloqueo de los canales de RyR2 y previeron la inducción de taquicardia polimórfica con el ejercicio físico en ratones, siendo significativamente más baja la potencia de la propafenona para bloquear los canales de RyR2. Clínicamente la propafenona (900 mg-día) previó las descargas eléctricas del desfibrilador automático en un joven de 22 años con TVPC que había sido refractario a la máxima dosis tolerada de β-bloqueadores y a la ganglio-ectomía simpática cardíaca bilateral. El verapamilo, la flecainida y la propafenona deberán ser valoradas como alternativas terapéuticas en los pacientes sintomáticos tratados con β-bloqueadores.
REFERENCIAS BIBLIOGRÁFICAS
1. Marks A, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia J Cell Physiol. 2002;190:1-6.
2. Reid DS, Tynan M, Braidwood L, Fitzgerald. Bidirectional tachycardia in a child. A study using His bundle electrography. Br Heart J. 1975;37:339-44.
3. Coumel P, Fidelle J, Lucet V, Attuel P, Bouvrain Y. Catecholamine induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: Report of four cases. Br Heart J. 1978;40(Suppl):28-37.
4. Dubner SJ, Gimeno GM, Elencwajg B et al. Ventricular fibrillation with spontaneous reversion on ambulatory ECG in the absence of heart disease. Am Heart J. 1983;105:691-3.
5. Cohen TJ, Liem LB, Hancock EW. Association of bidirectional ventricular tachycardia with familial sudden death syndrome. Ain J Cardiol. 1989;64:1078- 9.
6. De Paola AAV, Horowitz LN, Marques FB. Control of multiform ventricular tachycardia by propranolol in a child with no identifiable cardiac disease and sudden death. Am Heart J. 1990;119:1429-32.
7. Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512-9.
8. Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia (CPVT).Circulation. 2002;106:69-74.
9. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196-200.
10. Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from
11. Leite LR, Ponzi Pereira KR, Alessi SR, de Paola AAV. Catecholaminergic polymorphic ventricular tachycardia. An important diagnosis in children with syncope and normal heart. Arq Bras Cardiol. 2001;76:63-74.
12. Napolitano C, Priori SG. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4(5):675-78.
13. Fisher JD, Krikler D, Hallidie-Smith KA. Familial polymorphic ventricular arrhythmias: a quarter century of successful medical treatment based on serial exercise-pharmacologic testing. J Am Coll Cardiol. 1999;34:201522.
14. Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff JM, Vaksmann G, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005;42:863-70.
15. Aizawa Y, Komura S, Okada S, Chinushi M, Aizawa Y, Morita H, et al. Distinct U wave changes in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT). Int Heart J. 2006;47:3819.
16. Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:48590.
17. Jiang D, Wang R, Xiao B, Wang R, Choi P, Zhang L, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:117381.
18. George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA. Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res. 2006;98:8897.
19. George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:53140.
20. Liu N, Rizzi N, Boveri L. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learn from genetic diseases and transgenic mice? J Mol Cell Cardiol. 2009;46:149-59.
21. Di Barletta MR, Viatchenko-Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114:10129.
22. Viatchenko-Karpinski S, Terentyev D, Gyorke I, Terentyeva R, Volpe P, Priori SG, et al. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94: 471-7.
23. Tester DJ, Arya P, Will M, Haglund CM, Farley
24. Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, et al. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208-14.
25. Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e7782.
26. Nam GB, Burashnikov B, Antzclevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;11:2727-33.
27. Schwensen C. Ventricular tachycardia as the result of the administration of digitalis. Heart. 1922;9:199-204.
28. Stubbs WA. Bidirectional ventricular tachycardia in familial hypokalaemic periodic paralysis. Proc R Soc Med. 1976;69:2234.
29. Morita H, Zipes DP,
30. Berte B, Eyskens B, Meyfroidt G, Willems R. Bidirectional ventricular tachycardia in fulminant myocarditis. Europace. 2008;10:7678.
31. Rosenbaum MB, Elizari MV, Lazzari JO. The mechanism of bidirectional tachycardia. Am Heart J. 1969;78:2-4.
32. Baher AA, Uy M, Xie F. Bidirectional ventricular tachycardia:
33. Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, et al. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89:66-70.
34. Priori S, Schwartz PJ, Napolitano C. Risk Stratification in the Long-QT Síndrome. N Engl J Med. 2003;348:1866-74.
35. Zhang L, Benson D.W, Tristani-Firouzi M. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-Uwave patterns predict the KCNJ2 genotype. Circulation. 2005;111:2720-6.
36. Hayashi M, Denjoy I, Extramiana F. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426-34.
37. Fisher JD, Krikler D, Hallidie-Smith KA. Familial polymorphic ventricular arrhythmias. A quarter century of successful medical treatment based on serial exercisepharmacologic testing. J Am Coll Cardiol. 1999;34:2015-22.
38. Heidbuchel H, Corrado D, Biffi A, Hoffmann E, Panhuyzen-Goedkoop N, Hoogsteen J, et al. Recommendations for participation in leisure-time physical activity and competitive sports of patients with arrhythmias and potentially arrhythmogenic conditions. Part II: ventricular arrhythmias, channelopathies and implantable defibrillators. Eur J Cardiovasc Prev Rehabil. 2006;13:676-86.
39. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eurospace. 2006;8:746-837.
40. Wilde AA, Bhuiyan ZA, Crotti L, Facchini M, De Ferrari GM, Paul T, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358:2024-9.
41. Swan H, Laitinen P, Kontula K, Toivonen L. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol. 2005;16:1626.
42. Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, et al. Calcium channel blockers and beta blockers versus betablockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149-54.
43. Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:3803.
44. Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circulation: Arrhythmia and Electrophysiology. 2011;4:128-35.
Recibido: 30 de marzo del 2012.
Aprobado: 1 de junio del 2012.
Dra. Marleny Cruz Cardentey. Hospital Clinicoquirúrgico “Hermanos Ameijeiras”.

{kind=link}
{kind=link}