










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versão impressa ISSN 0864-0300
Rev Cubana Invest Bioméd vol.33 no.1 Ciudad de la Habana jan.-mar. 2014
ARTÍCULO DE REVISIÓN
Metilación del ADN: implicaciones en carcinogénesis
DNA methylation and implications in carcinogenesis
BSc. Diego Fernando Uribe Yunda,I,II MSc. Fabián M. Cortes ManceraI
I Instituto Tecnológico Metropolitano (ITM). Medellín, Colombia.
II Universidad de Antioquia (UdeA), Medellín-Colombia.
RESUMEN
El cáncer se ha constituido en uno de los principales problemas de la salud pública mundial, no solo por su casuística sino por la alta morbilidad y mortalidad asociadas; solo en 2008 se estimaron más de 12 millones de nuevos casos de cáncer diagnosticados, 7 millones de muertes y 25 millones de personas vivas con este padecimiento. A nivel celular y molecular, el cáncer se define como una alteración de los mecanismos que regulan la división celular. Entre estos mecanismos, la epigenética estudia los cambios heredables que afectan el patrón de la expresión génica, que no son consecuencia de alteraciones en la secuencia nucleotídica del gen (mutaciones), o de sus secuencias reguladoras (promotores). De estos cambios, la metilación del ADN es la mejor caracterizada, asociándose con el silenciamiento o sobreexpresión de genes claves en la regulación del inicio y la progresión del cáncer, como es el caso de los genes involucrados en la vía de señalización Wnt/β-catenina. Comprender los pasos implicados en el inicio y en el establecimiento de alteraciones en la expresión génica mediadas por fenómenos epigenéticos, permitirá desarrollar terapias dirigidas a componentes claves involucrados en este proceso. En el presente manuscrito se analizan algunos mecanismos epigenéticos, su efecto sobre la regulación de la expresión génica, y su papel en la carcinogénesis.
Palabras clave: cáncer, epigenética, metilación del ADN, Wnt/β-catenina.
ABSTRACT
Cancer has become one of the main public health problems worldwide, not only for its incidence, but also due to its high morbidity and mortality. Only in 2008, an estimated 12 million new cancer cases were diagnosed, with 7 million deaths and 25 million people living with the disease. On a cellular and molecular level, cancer is defined as an alteration in the mechanisms regulating cell division. Epigenetics is the study of inheritable changes affecting the gene expression pattern in these mechanisms, which are not a consequence of alterations in the nucleotide sequence of the gene (mutations) or its regulatory sequences (promoters). Among these changes, DNA methylation has been characterized most accurately. It has been associated with the silencing or overexpression of genes performing a key role in regulating the start and progress of cancer, such as the genes involved in the Wnt/β-catenin signaling pathway. Understanding the steps involved in the start and establishment of gene expression alterations mediated by epigenetic phenomena, will make it possible to develop therapies aimed at key components of this process. The present study is an analysis of some epigenetic mechanisms, their effect on gene expression regulation, and their role in carcinogenesis.
Key words: cancer, epigenetics, DNA methylation, Wnt/β-catenin.
INTRODUCCIÓN
El cáncer es una enfermedad compleja tanto en su desarrollo como en la forma en que se manifiesta de un individuo a otro; se caracteriza por la presencia de alteraciones en los mecanismos genéticos y/o epigenéticos que regulan la división celular, llevando a la proliferación celular descontrolada (alteraciones en supresores tumorales, genes de reparación, reguladores del ciclo celular, adhesión, diferenciación celular y apoptosis, entre otros). De esta manera, las funciones fisiológicas de los tejidos afectados se perturban, dando lugar a múltiples manifestaciones clínicas, en dependencia del órgano comprometido, el tamaño del tumor y de la propagación de las células cancerígenas en el organismo (metástasis).1
De acuerdo con lo estimado por la Agencia Internacional de Investigación en Cáncer (IARC), tan solo en 2008 se presentaron aproximadamente 12 400 000 nuevos casos, y una mortalidad asociada a cáncer de 7,6 millones en todo el mundo. Asimismo, en la región que comprende el Caribe, Centro y Suramérica, según PAHO (Pan American Health Organization), se registraron 906 008 casos y 542 051 muertes. Para Colombia en este mismo periodo fueron notificados 27 597 casos y una mortalidad de 16 674 individuos para ambos sexos, mientras que en Chile se presentaron 18 441 nuevos casos de cáncer y una mortalidad de 11 492 casos para ambos sexos (estos datos no incluyen los tipos de cáncer de piel diferentes a melanoma). De manera preocupante, la prevalencia estimada a 5 años es de 28 803 166 casos nuevos por año en el mundo.1,2
Algunos mecanismos implicados en la transformación celular han sido explorados, sin embargo persisten varias interrogantes de este proceso multietapas, particularmente en lo concerniente a las alteraciones epigenéticas y a la secuencia de estos eventos en la carcinogénesis. Según Ting y colaboradores la epigenética se refiere a "cambios heredables en el patrón de expresión génica, que no son consecuencia de alteraciones en la secuencia nucleotídica del gen".3 Estos cambios están dados por diferentes mecanismos, de los cuales se destacan la metilación del ADN, la impronta genómica y la modificación de las proteínas histonas, que en conjunto interfieren silenciando o sobreexpresando genes que desregulan el funcionamiento celular normal, favoreciendo el inicio y la progresión tumoral. 4-6
Entre los mecanismos antes mencionados, la metilación del ADN en sitios ricos en citosinas y guaninas (islas CpG) es un factor que se ha visto alterado en carcinogénesis. En las células tumorales, las islas CpG localizadas en los promotores de genes involucrados en carcinogénesis se encuentran frecuentemente hipermetiladas, evitando la expresión del gen correspondiente, y favoreciendo la pérdida de heterocigocidad; esto último corresponde a la pérdida de la expresión génica mediante el daño en el alelo de un gen en el que su otro alelo había sido previamente inactivado. De igual forma, en las células tumorales otras regiones del genoma pueden estar hipometiladas, llevando a inestabilidad cromosómica y aparición de eventos de translocaciones.3,7,8
Por esta razón se vienen estudiando las alteraciones epigenéticas en componentes de diferentes vías de señalización,9 como es el caso de la vía Wnt/beta-catenina (Wnt/β-catenina),10,11 la cual viene siendo explorada por su papel en procesos de adhesión celular, proliferación, diferenciación y carcinogénesis.12 En esta vía se han observado alteraciones epigenéticas en los genes APC (adenomatous polyposis coli), CDH1 (cadherin 1, type 1, E-cadherin), WIF1 (WNT inhibitory factor 1), DACT3 (dapper, antagonist of beta-catenin, homolog 3), SOX (SRY- sex determining región Y- box containing genes), miembros de la familia SFRP (secreted frizzled-related protein), DKK ( dickkopf homolog) y WNT (wingless-type MMTV integration site family).
En el presente manuscrito se describen los mecanismos asociados a alteraciones epigenéticas y su importancia en la regulación de la expresión génica, tomando como modelo algunas de las alteraciones epigenéticas reportadas en la vía Wnt/β-catenina.
VÍA Wnt/β-CATENINA
Transducción de señales y función de Wnt/β-catenina
La vía Wnt/β-catenina está involucrada en la embriogénesis, proliferación y en las interacciones célula-célula en tejidos. Como se observa en la figura 1a, β-catenina es el componente central en esta vía, al interactuar con la proteína E-cadherina y alfa-catenina acoplándose al citoesqueleto. En ausencia de transducción de señales por Wnt (figura 1b), β-catenina es fosforilada en los residuos serina-treonina del dominio amino-terminal por GSK3β (glycogen synthase kinase 3 beta) y CKI (casein kinase ), en complejo con APC y AXIN. Los residuos fosforilados son reconocidos por BTrCP (beta-transducin repeat containing protein) y por la ubiquitina ligasa E3, los cuales inducen la degradación de β-catenina por vía proteosomal.12
La vía Wnt/β-catenina ha sido objeto de numerosos estudios, porque su alteración ha emergido como un mecanismo preponderante en el desarrollo de neoplasias que presentan casuísticas importantes, como el cáncer de mama, el carcinoma colorrectal (CCR), cáncer gástrico y hepático; en 2008 para estas cuatro neoplasias a nivel mundial fueron reportados 4 288 609 nuevos casos y más de 2,5 millones de decesos relacionados con estas afecciones. Aunque el cáncer de mama fue el que aportó más al total de casos (1 345 155), seguido por el CCR (1 235 108), una mayor mortalidad estuvo asociada con el cáncer gástrico (737 419) y el hepático (695 726).2,10 En Latinoamérica y el Caribe, una tendencia similar fue observada ese mismo año, en el cual se registraron 272 750 nuevos casos de estas neoplasias y 158 503 fallecimientos asociados; para esta región el cáncer de mama correspondió a la neoplasia con mayor número de nuevos diagnósticos (114 898), mientras que el cáncer gástrico resultó con la más alta cantidad de defunciones relacionadas (54 308).2,10
Particularmente, en tumores del tracto gastrointestinal (TTG), que incluyen neoplasias originadas en esófago, estómago, hígado, sistema biliar (conducto y vesícula biliar), páncreas, intestino, colon y ano, la vía de señalización Wnt/β-catenina se encuentra frecuentemente activada.13 Según la IARC, en 2008 se presentaron 3 878 986 casos de TTG en el mundo, lo que corresponde al 30 % del total de tumores reportados ese mismo año, y 2 824 985 muertes asociadas a TTG, lo que corresponde al 37,3 % del total de muertes asociadas a cáncer en ese año (tabla 1).2 Entre los factores de riesgo de TTG, están el consumo de tabaco, las infecciones crónicas ( Helycobacter pylori, virus de la hepatitis B y hepatitis C, Trematodos hepáticos), alcohol, dieta, exposición ocupacional (compuestos del plomo, bifenilos policronilados, tetracloroetileno) y carcinógenos ambientales (aflatoxinas, tricloroetileno).1

En diversos tumores se ha descrito la activación de β-catenina independiente de Wnt, y se ha observado que esta activación se da por alteraciones tanto genéticas como epigenéticas, por lo que algunas de ellas serán discutidas.
Desregulación de la vía Wnt/β-catenina por alteraciones genéticas
Varias han sido las mutaciones reportadas en los genes que codifican para proteínas involucradas en esta vía de señalización. Algunas de estas mutaciones han sido halladas en el gen de β-catenina (CTNNB1, cadherin-associated protein), particularmente en las secuencias que codifican para los residuos fosforilables por GSK3β, lo cual conlleva a la estabilización citoplasmática de la proteína (fig. 1), su posterior translocación a núcleo y la transcripción de los blancos de Wnt;14 también han sido observadas mutaciones en AXIN 1 y AXIN 2.15 Asimismo, en diferentes neoplasias han sido reportadas mutaciones que inactivan el antagonista extracelular SFRP5 y mutaciones que activan constitutivamente el receptor de la vía, frizzled (Fz) y el correceptor LRP5 ( low density lipoprotein receptor-related protein 5).10 De forma llamativa, en diferentes casuísticas de CCR ha sido descrita la presencia de mutaciones y pérdida de heterocigocidad en el gen APC en más del 80 % de los casos,15 muy superior a lo observado en carcinoma hepatocelular (CHC), indicando la existencia de otros mecanismos en la desregulación de la vía en neoplasias del hígado.
Epigenética y carcinogénesis
Mecanismos epigenéticos
En las células eucariotas, existen diferentes formas para asegurar el adecuado empaquetamiento del ADN, como estrategia de regulación de la expresión génica. Entre estos se encuentran la metilación del ADN y la modificación de las histonas.3
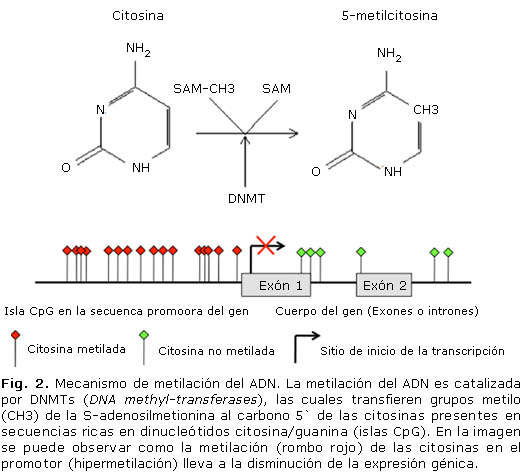
La metilación del ADN es catalizada por enzimas DNMTs (DNA methyl-transferases), las cuales transfieren grupos metilo (CH3) de la S-adenosilmetionina al carbono 5` de las citosinas presentes en islas CpG (fig. 2). Estos metilos quedan proyectados en el surco mayor del ADN y son reconocidos por proteínas denominadas MBDs (Methyl-CpG-binding domain proteins), las cuales interfieren con la unión de los activadores transcripcionales al ADN.4 A su vez, esta metilación puede promover la desaminación de las bases nitrogenadas, incrementando la absorción de luz UV por el ADN, intensificando su afinidad por carcinógenos, llevando a la formación de aductos y mutaciones.6
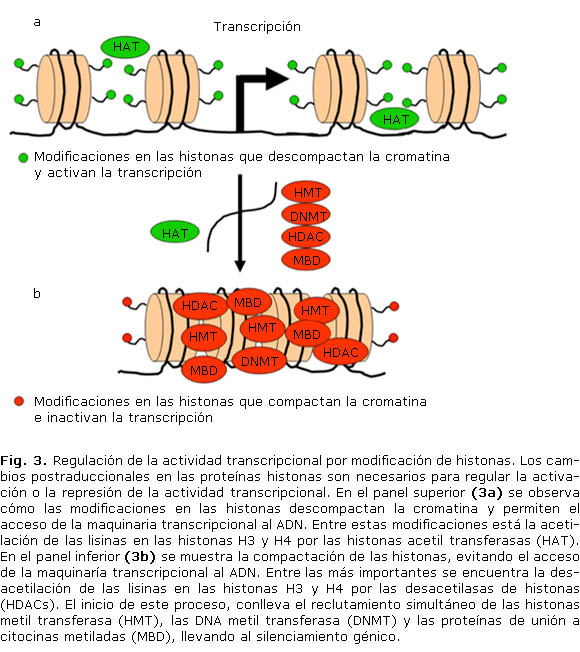
Por otro lado están las histonas (H1, H2A, H2B, H3 y H4), que corresponden a proteínas que se localizan en el núcleo, alrededor de las cuales el ADN se enrolla para favorecer la compactación la cromatina.16 Aunque la metilación del ADN es clave en la regulación transcripcional, modificaciones en las histonas‒ principalmente en las H3 y H4-, permiten el empaquetamiento de la cromatina y el subsecuente silenciamiento génico,3,6 estos mecanismos de regulación están frecuentemente alterados en el cáncer.17 En este contexto, las enzimas metiladoras de histonas, denominadas HMTs (Histone methyl-transferases), reclutan diferentes DNMTs a los promotores de los genes,18 las cuales a su vez reclutan enzimas desacetilasas (HDACs; histone deacetilases) y proteínas MBDs, bloqueando el acceso de la maquinaria transcripcional al promotor (fig. 3).19 Este modelo explica por qué la cromatina que está rodeando un gen transcripcionalmente activo es tan diferente de la que rodea un gen silenciado por mutilación,3,6,19 además sugiere que la adición de grupos metilo a las histonas se da en las fases iniciales del silenciamiento transcripcional, evento que es intensificado posteriormente por la metilación del ADN;17,20 en la tabla 2 se resumen las modificaciones más relevantes en proteínas histonas.
Metilación del ADN y su relación con la carcinogénesis
A partir de 1983, cuando se describió la alteración en el patrón de metilación en células tumorales,21 surgió la pregunta de si los mecanismos epigenéticos estaban involucrados en la carcinogénesis.5 Para estudiar esta relación, Holm y colaboradores generaron un modelo animal capaz de producir células embrionarias en las que se podía controlar el nivel de DNMTs tipo 1 (Dnmt1), logrando establecer ratones que no mantenían la metilación del ADN en aquellos locus donde estaban ubicados oncogenes y supresores tumorales, proporcionando el "primer paso" para la formación tumoral.22 Igualmente, Yamada y colaboradores demostraron que la hipometilación global del genoma promueve el inicio del desarrollo tumoral en colon, hígado e intestino delgado, en ratones con solo un alelo funcional del gen que codifica para APC (fig.1).23
En un estudio realizado por Igarashi y colaboradores en 2010, se demostró que la metilación del marcador génico LINE-1 estaba inversamente correlacionada con el tamaño y con el índice mitótico del tumor, parámetros que fueron utilizados para determinar la agresividad en una serie de 85 tumores estromales gastrointestinales. De esta manera, los investigadores observaron que los niveles de metilación de LINE-1 eran significativamente menores en los tumores de alto riesgo que en los de riesgo bajo o intermedio.24
Asimismo, la hipometilación de LINE-1 ha sido asociada con un pronóstico pobre en carcinoma de próstata,25 colon,26 ovario27 y leucemia mieloide crónica.28 En conjunto estos resultados postulan la metilación de LINE-1 como un marcador para evaluar la aparición y progresión de estos tumores y su potencial metastático,29 por lo que son necesarios estudios adicionales que permitan determinar exactamente cuál es el papel de la hipometilación en cada una de estas enfermedades.26
Alteraciones epigenéticas en la vía Wnt/β-catenina y cáncer
Como se muestra en la figura 1b, APC es un componente esencial del complejo encargado de la fosforilación y degradación de β-catenina cuando no hay activación de la vía. En este contexto, E-cadherina se puede unir directamente a β-catenina, asegurando la adhesión celular y evitando su translocación a núcleo, y la expresión de genes blancos;12 tanto APC como E-cadherina ayudan a mantener el control de la vía, sin embargo en algunas neoplasias se han observado alteraciones epigenéticas en estos genes.
En este sentido, la hipermetilación de los promotores de APC y CDH1 (el gen que codifica para E-cadherina) ha sido descrita en CHC, favoreciendo la estabilización de β-catenina en este cáncer.30,31 Katoh y colaboradores evaluaron retrospectivamente la metilación de promotores de 9 supresores tumorales en 60 muestras de CHC y encontraron que un alto grado de metilación se correlacionaba con la inestabilidad cromosómica de los tumores. Estos resultados sugieren que las alteraciones en carcinogénesis, pueden darse por un evento epigenético inicial y que la metilación de APC y CDH1 (88,3 y 43,3 %, respectivamente) puede ser un suceso frecuente en CHC.30 De igual forma Prasad y colaboradores observaron hipermetilación de los promotores de CDH1 (50 %) y APC (22 %) en casos de carcinoma ductal invasivo de mama, lo cual se correlacionó con ausencia de las proteínas correspondientes. Además, la localización nuclear de β-catenina se observó en 57 % y 50 % de los tumores que presentaban metilación en CDH1 y APC, lo que se asoció con expresión de marcadores de transición epitelial; estos resultados sugieren que la activación de Wnt/β-catenina puede darse por eventos epigenéticos, favoreciendo además la pérdida de la adhesión célula-célula.32
Otro mecanismo implicado en la activación de β-catenina es el silenciamiento por hipermetilación de los genes antagonistas de Wnt (WIF1, DKK3 y SFRP) (fig. 1a), los cuales evitan la unión de las moléculas Wnt a los receptores Fz y al correceptor LRP5/6.33 Taniguchi y colaboradores demostraron el restablecimiento de la expresión de WIF1 en líneas celulares gastrointestinales, al tratarlas con 5-aza-2´deoxicitidina (inhibidor de las DNMTs) y con tricostatina A (inhibidor de las HDACs), sugiriendo que la metilación del ADN y la desacetilación de histonas están involucradas en este silenciamiento. Adicionalmente, observaron reducción en la expresión de WIF1 en 77,8 % de los tejidos tumorales, demostrando relación con hipermetilación. Finalmente, al transfectar células que no expresaban WIF1 con un constructo que contenía este gen, se observó inhibición significativa en la formación de colonias, en la proliferación celular y la actividad transcripcional dependiente de TCF. Estos resultados sugieren que el silenciamiento de WIF1 por fenómenos epigenéticos es un mecanismo importante para la activación de la vía Wnt/β-catenina,34 evidenciando además el potencial antitumoral que tiene la terapia basada en agentes demetilantes del ADN e inhibidores de HDACs. Adicionalmente, Veeck y colaboradores describieron que la hipermetilación de los promotores de WIF1 (63,3 %) y DKK3 (61,3 %) puede llegar a ser frecuente en carcinoma de mama, y propusieron la evaluación del estado de metilación como un biomarcador pronóstico, ya que aquellos pacientes con metilación (M) del promotor de DKK3 tuvieron un desenlace menos favorable comparado con quienes no presentaban metilación (UM) (sobrevida a 10 años: 54 % M vs. 97 % UM, y sobrevida libre de enfermedad a 10 años: 58 % M vs. 78 % UM).35
En otro trabajo, Kaur y colaboradores demostraron que SFRP1 está frecuentemente silenciado por hipermetilación en casos de cirrosis hepática y CHC; al restaurar la expresión de SFRP1 en células de CHC que no expresaban la proteína; estos investigadores observaron potenciación en la inducción de apoptosis con una disminución significativa del crecimiento celular, indicando que el silenciamiento de SFRP1 por hipermetilación aumenta el potencial tumorigénico de las células, por su resistencia a la apoptosis y mayor capacidad proliferativa. Además, la hipermetilación de SFRP1 puede postularse como un potencial biomarcador para la detección temprana del CHC y un potencial blanco terapéutico.36
En nuestro grupo se están realizando ensayos para determinar la localización subcelular de β-catenina en líneas celulares de cáncer de hígado con diferente "background" genético. De este modo, se ha observado que en las células HepG2, β-catenina presenta un fuerte marcaje en membrana y citoplasma, observando incluso algunas células con localización nuclear de la proteína, lo cual puede ser explicado por la detección de los aminoácidos que codifican para los sitios de fosforilación por GSK3β y CKI y/o por la hipermetilación de alguno de los antagonistas extracelulares de Wnt (datos no publicados).
Recientemente fue descrita la familia de proteínas DACT3, las cuales modulan la vía de Wnt/β-catenina al interactuar con la fosfoproteína DVL (Dishevelled),37 evitando de esta manera la inactivación de GSK3β, mediada por DVL.38 Jiang y colaboradores observaron que la expresión de DACT3 en líneas celulares de carcinoma de colon es baja, contrario a lo evidenciado en células normales del epitelio intestinal. Adicionalmente, tanto la reconstitución de la expresión de DACT3 utilizando agentes demetilantes e inhibidores de HDACs, como la expresión exógena de la proteína, desencadena una degradación de DVL2, reduciendo la concentración intracelular de β-catenina e incrementando la apoptosis, aun en líneas celulares con mutaciones en APC. Los resultados de este estudio, permitieron identificar otro mecanismo de regulación de la vía Wnt/β-catenina y sugirieron que el tratamiento basado en agentes que modulan los mecanismos epigenéticos, constituye una interesante alternativa terapéutica.39
Aunque se ha demostrado que la desregulación de la vía de señalización Wnt/β-catenina es preponderante en el cáncer, son muchos los genes susceptibles de ser alterados por mecanismos epigenéticos y que juegan un papel en la carcinogénesis; la identificación, tanto de los mecanismos como de los actores involucrados, permitirá el diseño de nuevas estrategias para el tratamiento del cáncer, como es el caso de agentes demetilantes e inhibidores de enzimas que modifican las histonas.
CONCLUSIONES
La importancia biológica de la epigenética en la regulación de la expresión génica y su papel en la carcinogénesis ha sido evidenciada por numerosos estudios. Recientemente, se ha observado que la epigenética puede tener un papel aún más importante que el atribuido inicialmente a este mecanismo, ya que se ha demostrado que estas alteraciones pueden estar involucradas no solo en la progresión sino que pueden constituir el mecanismo inicial en el proceso de la carcinogénesis, favoreciendo la aparición y el establecimiento de alteraciones genéticas y, de esta manera, la transformación celular. Entre estos mecanismos, la metilación ha sido el mejor caracterizado y se ha evidenciado su implicación en el silenciamiento de genes claves en el inicio y la progresión del cáncer. Razón por la cual, es imperiosa la necesidad de diseñar estudios que permitan entender los mecanismos moleculares implicados en el inicio y en el establecimiento del silenciamiento génico mediado por fenómenos epigenéticos y, de esta manera, identificar potenciales blancos terapéuticos. Adicionalmente, la detección de la metilación de los promotores de los genes puede utilizarse como un marcador molecular importante para ayudar a determinar el riesgo de desarrollar cáncer y de esta manera constituirse como un marcador diagnóstico temprano y adicionalmente como un potencial marcador pronóstico.
Agradecimientos
Los autores agradecen a la Dirección de Investigaciones del Instituto Tecnológico Metropolitano (ITM), institución universitaria adscrita a la Alcaldía de Medellín (P10242).
REFERENCIAS BIBLIOGRÁFICAS
1. Boyle P, Levin B, Editores. World Cancer Report 2008. Lyon, Francia: International Agency for Research on Cancer; 2008:
2. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 v2.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10 [Internet]. Lyon, France: International Agency for Research on Cancer; 2010. Available from: http://globocan.iarc.fr [accessed on 24/10/2012].
3. Ting AH, McGarvey KM, Baylin SB. The cancer epigenome - components and functional correlates. Genes Dev. 2006;20:3215-31.
4. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89-97.
5. Feinberg AP The epigenetics of cancer etiology. Semin Cancer Biol. 2004;14:427-32.
6. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(S1):S4-S11.
7. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286-98.
8. Esteller M. Epigenetics in Cancer. N Engl J Med. 2008;358:1148-59.
9. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Mutations and Deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascades. Oncotarget. 2012;3(9):954-87.
10. Ying Y, Qian T. Epigenetic disruption of the WNT/β-catenin signaling pathway in human cancers. Epigenetics. 2009;4:5,307-12.
11. Aguilera O, Muñoz A, Esteller M, Fraga MF. Epigenetic alterations of the Wnt/beta-catenin pathway in human disease. Endocr Metab Immune Disord Drug Targets. 2007;7(1):13-21.
12. Brembeck FH, Rosário M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of β-catenin. Curr Opin Genet Dev. 2006;16:51-9.
13. White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142(2):219-32.
14. Morin PJ, Sparks AB, Korinek V, Barrer N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787-90.
15. Ishizaki Y, Ikeda S, Fujimori M, Shimizu Y, Kurihara T, Itamoto T, et al. Immunohistochemical analysis and mutational analyses of β-catenin, axin family and APC genes in hepatocellular carcinomas. Int J Oncol. 2004;24(5):1077-83.
16. Khare SP, Habib F, Sharma R, Gadewal N, Gupta S, Galande S. Hlstome- a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 2012 Jan;40 s(Database issue):D337-42.
17. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107-116.
18. Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, et al. Role of Histone H3 Lysine 27 Methylation in X Inactivation. Science. 2003;300:131-5.
19. Fahrner JA, Eguchi S, Herman JG, Baylin SB. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002;62:7213-8.
20. Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, et al. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell. 2003;3:89-95.
21. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143-53.
22. Holm TM, Jackson-Grusby L, Brambrink T, Yamada Y, Rideout WM, Jaenisch R. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell. 2005;8(4):275-85.
23. Yamada Y, Jackson-Grusby L, Linhart H, Meissner A, Eden A, Lin H, et al. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. PNAS. 2005;2(38):13580-5.
24. Igarashi S, Suzuki H, Niinuma T, Shimizu H, Nojima M, Iwaki H, et al. A Novel Correlation between LINE-1 Hypomethylation and the Malignancy of Gastrointestinal Stromal Tumors. Clin Cancer Res. 2010;16:5114-23.
25. Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, et al. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol. 2007; 211:269-277.
26. Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Chan AT, Schernhammer ES, et al. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J Natl Cancer Inst. 2008;100:1734-8.
27. Pattamadilok J, Huapai N, Rattanatanyong P, Vasurattana A, Triratanachat S, Tresukosol D, et al. LINE-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int J Gynecol Cancer. 2008;18:711-7.
28. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Cervantes F, Sanchez J, Garate L, et al. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene. 2005;24:7213-23.
29. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239-59.
30. Katoh H, Shibata T, Kokubu A, Ojima H, Fukayama M, Kanai Y, et al. Epigenetic Instability and Chromosomal Instability in Hepatocellular Carcinoma. Am J Pathol. 2006;168(4):1375-83.
31. Yang B, Guo M, Herman JG, Clark DP. Aberrant Promoter Methylation Profiles of Tumor Suppressor Genes in Hepatocellular Carcinoma. Am J Pathol. 2003;163(3):1101-7.
32. Prasad CP, Mirza S, Sharma G, Prashad R, DattaGupta S, Rath G, et al. Epigenetic alterations of CDH1 and APC genes: relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast. Life Sci. 2008;83(9-10):318-25.
33. Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003 Jul 1;116(Pt 13):2627-34.
34. Taniguchi H, Yamamoto H, Hirata T, Miyamoto N, Oki M, Nosho K, et al. Frequent epigenetic inactivation of Wnt inhibitory factor-1 in human gastrointestinal cancers. Oncogene. 2005;24:7946-52.
35. Veeck J, Wild PJ, Fuchs T, Schüffler PJ, Hartmann A, Knüchel R, et al. Prognostic relevance of Wnt-inhibitory factor-1 (WIF1) and Dickkopf-3 (DKK3) promoter methylation in human breast cancer. BMC Cancer. 2009 Jul;(9):217.
36. Kaur P, Mani S, Cros MP, Scoazec JY, Chemin I, Hainaut P, et al. Epigenetic silencing of sFRP1 activates the canonical Wnt pathway and contributes to increased cell growth and proliferation in hepatocellular carcinoma. Tumour Biol. 2012;33(2):325-36.
37. Fisher DA, Kivimäe S, Hoshino J, Suriben R, Martin PM, Baxter N, et al. Three Dact gene family members are expressed during embryonic development and in the adult brains of mice. Dev Dyn. 2006;235(9):2620-30.
38. Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao Y, Wu D, et al. Direct Binding of the PDZ Domain of Dishevelled to a Conserved Internal Sequence in the C-Terminal Region of Frizzled. Mol Cell. 2003;12:1251-60.
39. Jiang X, Tan J, Li J, Kivimäe S, Yang X, Zhuang L, et al. DACT3 is an epigenetic regulator of Wnt/β-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell. 2008;13(6):529-41.
Recibido : 1ro. de octubre de 2013.
Aprobado: 20 de diciembre de 2013.
Fabián M. Cortés-Mancera. Instituto Tecnológico Metropolitano (ITM), Calle 73 No 76A -354 Vía al Volador - Medellín, Colombia. CO-549 59. Tel: 0057+4+440 52 91. Cel: 0057+4+440 52 91 Fax: 0057+4+440 51 03. Correo electrónico: fabiancortes@itm.edu.co

{kind=link}
{kind=link}
{kind=link}
{kind=link}