










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
La insuficiencia cardiaca (IC) es una de las principales causas de hospitalización y mortalidad a nivel mundial. A pesar de los avances terapéuticos, los índices de incidencia y prevalencia son elevados.1 La IC se define como un síndrome caracterizado por la incapacidad de proveer las necesidades metabólicas del organismo, en ella se incluyen la hipertensión y la isquemia miocárdica.2
Como un factor predictivo la hipertrofia ventricular (HV) se manifiesta tempranamente como parte de la respuesta cardiaca a la sobrecarga crónica, que, por lo general, no suele diagnosticarse oportunamente. La sobrecarga crónica involucra una serie de cambios anatomorfofisiológicos del miocardio (hipertensión arterial, sobrecarga hemodinámica, remodelación miocárdica, hipertrofia ventricular, insuficiencia cardiaca) y es el primer paso para llegar a la falla cardiaca.1,3
Se ha descrito la participación de diversas moléculas reguladoras de los mecanismos moleculares en los procesos mecánicos, lo que contribuye al daño al miocardio y a la generación de la IC. Dentro de los mecanismos biológicos que participan en la IC se encuentran múltiples alteraciones: en los circuitos de óxido-reducción, el manejo intracelular de Ca2+, la disfunción mitocondrial y la presencia de mutaciones en proteínas específicas.1,3
El presente trabajo se enfoca a la revisión e integración de información de las biomoléculas participantes en los mecanismos de acción que contribuyen a la generación de la IC. Para efectuar la presente revisión se realizó la búsqueda de información en bases de datos electrónicas.
Participación de las especies reactivas de oxígeno (ERO) en la actividad cardiaca
El nivel de oxígeno en el corazón no es determinante para la expresión génica miocárdica, pero sus productos sí tienen relevancia, tal es el caso de las especies reactivas de oxígeno (ERO), como el óxido nítrico (ON), el cual está involucrado en la determinación del tono vascular, la contractilidad miocárdica, además de ser antitrombótico, vasodilatador, antiinflamatorio, antioxidante e inhibidor del crecimiento celular.3,4
Las ERO pueden participar de forma benéfica en la señalización intracelular o bien pueden inducir un daño celular irreversible y desencadenar el mecanismo de apoptosis en los cardiomiocitos. Esto se logra directamente al interactuar con los lípidos celulares produciendo principalmente peroxidación y un subsecuente daño a la membrana celular.5,6Además, debido a su capacidad mutagénica, estas moléculas interactúan con diversas proteínas provocando inactivación y desnaturalización de algunas enzimas.6,7
Efecto de la angiotensina II en la insuficiencia cardiaca
Se ha sugerido que la angiotensina II (Ang II) es un factor predisponente al desarrollo de IC. La Ang II se une selectivamente a su receptor AT1,está asociado a proteínas G, y activauna cascada de eventos que involucran la producción de O2 y su conversión a H2O2 y OH- por efecto de la enzima superóxido dismutasa, estas son las mediadoras de la activación de una serie de proteincinasas conocidas como MAPKs (Fig.1). La activación de MAPK provoca hipertrofia miocárdica o a mecanismos apoptoticos.8,9
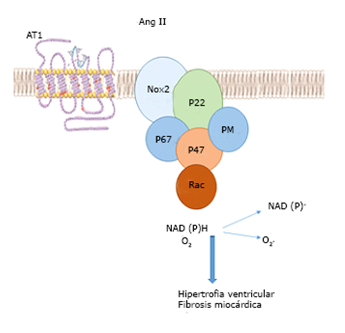
La Ang II actúa sobre los receptores AT1, y estos, a su vez, lo hacen sobre Nox2 en los cardiomiocitos, provocando hipertrofia ventricular a través de la NADP (H).
Los niveles de Ang II se elevan a consecuencia de la disminución del volumen/minuto o por estimulación simpática activando al sistema renina angiotensina (SRA). Pero puede suceder que el sistema local SRA esté activado selectivamente en el corazón sobrecargado y que la Ang II circulante permanezca en niveles normales.9
La activación a largo plazo del SRA cardiaco conduce a que se desencadene la hipertrofia cardiaca, que es independiente de los niveles sistémicos de la hormona. Así mismo, produce hipertrofia del músculo liso vascular, mediada por radicales libres, los que activan cascadas de señalización y tienen efectos mitogénicos.
Un aumento de estrés de la pared ventricular activa al SRA y, por consiguiente, se incrementa la Ang II, responsable de mayor rigidez cardiaca.9 Se ha sugerido que las concentraciones elevadas de esta hormona en relación a tasas correspondientes de excreción de sodio (Na+) se corresponden con una HV exagerada en la hipertensión arterial, puesto que a su vez estimulan la expresión local de aldosterona, conduciendo así a una síntesis de colágeno de tipo 1 y procolágeno de tipo III. Todo ello favorece los cambios anatomo-estructurales del ventrículo izquierdo en respuesta a un aumento de carga.9,10
Por su parte, la oxidasa NADPH es relevante en el desarrollo de HV inducida por Ang II, en forma independiente de los cambios de la presión arterial (ya que promueve el estrés oxidativo).
Dinamia de las proteínas G en la IC
Por otro lado, las proteínas G son una familia de la cual se sugiere que las subunidades participan en mecanismos de IC: la Gs (stimulating), la Gi (inhibitory) y la Gq. Esta última estimula la fosfolipasa C y por ese intermedio al difosfatidilinositol, a partir del cual se forma inositol trifosfato (IP3) y diacilglicerol (DAG). El IP3 interviene en el movimiento y liberación del calcio (Ca2+) intracelular, mientras que el DAG activa la proteína-quinasas C (PkC), el que a su vez actúa por medio de una quinasa de la proteína serina/treonina, desencadenando la cascada de la proteína quinasa activada por mitogenos (MAPK). Esta vía de señalización también lleva a la hidrólisis del difosfatidilinositol y a la formación por DAG de PKC, activándose luego la fosfolipasa A y quinasas de tirosina.8,9
Las proteínas G acopladas al receptor juegan un papel muy importante en la señalización que induce HV. De forma particular, la sobreexpresión de receptores acoplados a la Gq en ratones transgénicos desencadena la HV, apoptosis e IC. Los receptores acoplados a la proteína G (GPCR) son capaces de activar la MAPK y, en ciertas condiciones, llevan a una respuesta mitogénica. La superfamilia MAPK incluye tres vías mayores: la quinasa extracelular regulada por señal (ERK)1/2 y dos vías de PKs activadas por estrés: c-Jun NH2-terminal kinase (JNK) y p38 MAPK. La activación de las MAPK por factores de crecimiento, citoquinas y estrés celular media selectivamente una variedad de respuestas celulares que van desde la diferenciación hasta mecanismos apoptóticos.10
Otros factores como el TGF y el IGF-1, a través de la Gq, confluyen hacia la MAPK. El IGF-1 es un péptido de 70 aminoácidos, con propiedades esenciales de factor de crecimiento para la proliferación y diferenciación celular durante el desarrollo. La activación de las proteína quinasa (PK) precede al aumento de la expresión del gen específico y a la síntesis proteica. Cuando son activadas las MAPK se translocan al núcleo donde hay numerosos factores de transcripción, quienes regulan la inducción de genes que determinan la respuesta biológica celular, incluyendo la hipertrofia.9
Akt una protein cinasa no específica
Otra molécula implicada en la generación de la IC es Akt, una serina-treonina quinasa involucrada en el desarrollo de la HV y en la contractilidad. Es el homólogo humano del oncogén viral v-Akt (retrovirus Akt 8) y está relacionado con proteínas quinasas A (PKA) y C (PKC) en seres humanos. Hasta ahora se ha propuesto la presencia de tres isoformas conocidas, derivadas de distintos genes: Akt1/PKBα, Akt2/PKBβ y Akt3/PKBγ.11
En modelo murino se ha visto que la sobreexpresión de Akt induce hipertrofia cardiaca, con un aumento significativo en el tamaño de los miocitos y la formación de HV concéntrica. Los ratones transgénicos con sobreexpresión de Akt también mostraron un marcado aumento de la contractilidad, donde la mecano-transducción estaría regulada por el ciclo cardiaco.11
Activación de NF-kB en pacientes con IC
El factor de transcripción NF-kB ha sido involucrado como participante causal en las respuestas de hipertrofia miocítica y es regulado por el TNF-α. Los pacientes con IC muestran niveles sanguíneos de TNF-α elevados y activación del NF-kB, el cual está retenido en el citoplasma ligado a la IKBs, esta última, un inhibidor citoplasmático del factor. Cuando hay estimulación se fosforila el IKBs por acción del complejo IKK. El NF-kβ al translocarse al núcleo se une a ADN a través de un dominio Rel, activando genes de respuesta inflamatoria e inmunológica, como el TNF-α, IKBa, angiotensinógeno, IL-2 e IL-8. Además interviene en el proceso apoptótico.12
Un vasoconstrictor en IC
Por su parte la endotelína-1 es un vasoconstrictor que actúa a través de receptores acoplados a la proteína G (GPCRs), principalmente de la subfamilia Gq/11,la queactiva a la PLC, formando IP3 y DAG; el DAG activa las formas delta y epsilon de PKCl. Posteriormente,se activa la estimulación de la MAP quinasa y la precipitación de la cascada ERK por la GPCRs. Como resultado de ello, el grado de dilatación frecuentemente excede la capacidad de hipertrofia por el consiguiente adelgazamiento de la pared ventricular.10,12
Calcio, un segundo mensajero involucrado en la IC
El mensajero más importante en el músculo cardiaco es el Ca2+, ya que participa en diversos procesos celulares como la expresión génica, la diferenciación y la apoptosis; además de ser componente indispensable del proceso de contracción. Parte de la disfunción del corazón en la IC se podría explicar por un aumento de la estimulación simpática y la concomitante liberación de catecolaminas, que induce la activación de los receptores β-adrenérgicos. Sin embargo, en los pacientes con IC crónica esta respuesta no se efectúa.
La contracción en los cardiomiocitos es disparada por el Ca2+, que ingresa desde el líquido extracelular (LEC) a través de canales iónicos tipo L, presentes en el sarcolema y cuya apertura es dependiente de voltaje, es decir, se abren durante la fase 2 ode meseta del potencial de acción (PA), cuando la membrana está despolarizada. El Ca2+ que ingresa provoca la liberación de más Ca2+ a partir de las reservas existentes en el retículo sarcoplásmico (RS), mecanismo conocido como liberación de Ca2+ inducida por Ca2+.
La salida de estas reservas ocurre a través de los llamados receptores de rianodina (RyR2), que son proteínas dispuestas en la membrana del RS, las que también funcionan como canales para Ca2+. Una vez que el Ca2+ aumenta en el citosol, interactúa con los miofilamentos provocando la activación de los puentes cruzadosy, con ello, la contracción (Fig. 2).11,12

La relajación del músculo cardiaco (lusiotropismo) está mediada por la recaptura del Ca2+ al RS, así como por la expulsión del ion hacia el LEC. Una ATPasa, conocida como la SERCA2a, es la proteína transportadora encargada de realizar esta recaptura en contra del gradiente de concentración. Las proteínas NCX1 y una Ca2+ ATPasa del sarcolema expulsan el Ca2+ hacia el LEC.
La actividad de los RyR2 es finamente modulada por múltiples reguladores. Uno de ellos, la calstabina 2, durante la diástole, se une a los RyR2 y los mantiene cerrados. En la IC, cuando los RyR2 son fosforilados por la proteína quinasa A (PKA), como resultado de la sobreestimulación adrenérgica, la calstabina se separa de los RyR2 y el Ca2+ sale del RS, en mayor proporción. Esta salida aumentada tiene 2 consecuencias: la generación de posdespolarizaciones tardías que pueden disparar una taquicardia ventricular y provocar la muerte súbita, y la disminución de las reservas de Ca2+ del RS, debido a que aumenta la proporción de este ion que es expulsado hacia el LEC y disminuye la que es recapturada en el RS. Lo anterior ocurre debido a que los transportadores NCX1 se encuentran muy activos y, en cambio, las SERCA2a están deprimidas.12
La actividad de la SERCA2a es regulada por el fosfolamban: una proteína ubicada en la membrana del RS, que al interactuar con ella la inhibe, pero cuando el fosfolamban está fosforilado, dicha inhibición pierde importancia.12
En la IC se reduce la fosforilación del fosfolamban, lo que contribuye a la disminución de la recaptura del Ca2+ en el RS. El Ca2+ además de cumplir una función en la contracción y relajación de los miocitos, también es importante como segundo mensajero en diferentes vías de señalización, dentro de las cuales están aquellas que se relacionan con la remodelación cardiaca.
Los cardiomiocitos son células diferenciadas que han perdido la capacidad de división, por lo tanto, un aumento en la masa cardiaca está básicamente determinado por un crecimiento celular. El problema que se presenta durante este crecimiento es que también ocurren alteraciones específicas en la expresión genética y en su fenotipo, algunas de tipo adaptativo, como ocurre con la expresión de genes que codifican para péptidos natriuréticos, para proteínas contráctiles fetales y otras de carácter no adaptativo, como sucede con la fibrosis.
Los cambios en el tamaño de la cavidad y en la estructura, además de incrementar la tensión sobre las paredes del corazón insuficiente, también deprimen su desempeño mecánico y pueden aumentar la regurgitación a través de la válvula mitral.
Péptidos participantes en IC
En la IC se observa un incremento en los niveles sanguíneos del péptido atrial natriurético (PAN) y del péptido cerebral natriurético (PCN). Ambos son producidos y secretados por el corazón ante un aumento de la distensión de las cavidades cardiacas; el PAN por los miocitos atriales y el PCN por los miocitos ventriculares. El primero parece secretarse principalmente cuando se producen cambios agudos y, el segundo, ante los cambios crónicos.
Los péptidos natriuréticos poseen varias acciones: tienden a inhibir al sistema simpático y al SRA, facilitan la natriuresis y la diuresis, disminuyen las resistencias periféricas, relajan el músculo liso y tienden a contrarrestar la acción de los mensajeros químicos que producen la remodelación cardiaca.
En principio, todas las acciones de los péptidos natriuréticos estarían fisiológicamente justificadas, pues tienden a compensar los efectos deletéreos ocasionados por la activación de sistemas como el simpático y el SRA.
Si bien es cierto muchos de los mecanismos compensadores presentados en esta revisión terminan deteriorando aún más la función cardiaca, este no sería el caso de los péptidos natriuréticos, los cuales participan disminuyendo tanto la poscarga como la precarga, que están elevadas debido al círculo vicioso que se presenta por la activación neurohumoral. Sin embargo, con el transcurso de la enfermedad tiende a atenuarse su liberación y a presentarse una regulación a la baja de sus receptores.8,12
Hipertrofia patológica
Los cambios moleculares observados durante el proceso desde la hipertrofia patológica se asemejan a los observados durante el desarrollo cardiaco fetal, dada la expresión de proteínas contráctiles fetales, y por lo tanto, la hipertrofia cardiaca se describe a menudo como un proceso acompañado por la reactivación de un “programa genético fetal” que puede involucrar adaptaciones saludables ante el estrés.
En este ámbito, se piensa que la activación del “programa genético fetal” tiene un papel importante en la remodelación cardiaca, además de que los marcadores moleculares que lo siguen podrían servir como biomarcadores clínicos siendo útiles a la progresión de la IC, como la proteína NT-proBNP, derivada de la activación del gen NPPB humano.13
Estudios recientes sugieren que la interrupción de la biogénesis mitocondrial es un cambio observado en estadios prematuros de la IC y que está involucrado en su evolución. El uso terapéutico de inhibidores de AMPK y el uso de NO/cGMP y resveratrol pueden facilitar la estimulación de la producción de nuevas mitocondrias,esto ha sido observado en ratones.13) Sin embargo, su funcionamiento en humanos aun es controversial, aunque las terapias actuales se enfocan en un tratamiento sintomático y en la inhibición del sistema renina angiotensina aldosterona (SRAA), estos recientes descubrimientos promueven un cambio en el enfoque del tratamiento dirigido a la estimulación directa de la biogénesis mitocondrial, reactivando la función de los cardiomiocitos.13,14

