










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Medicina General Integral
versão impressa ISSN 0864-2125versão On-line ISSN 1561-3038
Rev Cubana Med Gen Integr v.22 n.3 Ciudad de La Habana jul.-set. 2006
Síndrome de Noonam
Emilia Yamilka Valdés Macola,1 Berta Lidia Acuña Montero2 y Edilberto Fernández Cumbá1
Resumen
Se describe el caso de un paciente del sexo masculino de 19 años con síndrome de Noonam, caracterizado por deformidad del tórax y malformaciones menores descritas en la cara, dispensarizado por el Consultorio no. 1 perteneciente al Centro Médico de la Universidad de las Ciencias Informáticas, en el municipio La Lisa , Ciudad de La Habana. Este paciente en la actualidad tiene seguimiento en el servicio de neumología del Hospital Clínicoquirúrgico Hermanos Ameijeiras.
Palabras clave: Síndrome de Noonam, adolescente, atención primaria, dispensarización.
El síndrome de Noonam fue descrito en 1963 por Noonam y Ehmke en pacientes con estenosis valvular pulmonar, y se asocia a la baja estatura, el hipertelorismo y el retardo mental moderado, entre otras alteraciones. Es una entidad genética de herencia autosómica dominante.1
Los niños afectados de uno u otro sexo han recibido varias denominaciones: fenotipo Turner con cromosomas normales, XY con fenotipo Turner (varones), XX con fenotipo Turner (hembras), síndrome de Ullrich, fenotipo de Turner familiar, síndrome seudoturner y síndrome de Noonam.2
Entre las anomalías más comunes figuran la baja talla, pterigium colli, pectus carinatum o pectus excavatum, cúbito valgo, hipertelorismo, epicantus, hendidura palpebral antimongoloide, ptosis, micrognatia y anormalidades de los pabellones auriculares, asociadas con menor frecuencia a lesiones vertebrales.3
Se trata de la presentación de un caso de síndrome de Noonam, diagnosticado desde su infancia, con posterior seguimiento después de la dispensarización que se realizó en el Consultorio no. 1 perteneciente al Centro Médico de la Universidad de las Ciencias Informáticas.
El tórax es especial, pues además del ensanchamiento mamelonar, un pectus carinatum se superpone a un pectus excavatum. Este último, en experiencias referidas, está a menudo aislado. Las malformaciones encontradas en ecografía prenatal son interesantes: un higroma quístico cervical, un hidramnios y/o una anasarca feto-placentaria, deben hacer pensar en un síndrome de Noonam en una familia de riesgo. Si se trata de la gestación de una niña, el cariotipo descarta un síndrome de Turner.
Al nacimiento una observación detallada puede orientar el diagnóstico, pues se evidencia un exceso de piel en la nuca, una anasarca, un linfedema difuso y un distress respiratorio, que junto a un quilotórax y una cardiopatía congénita son reveladores de la presencia de este síndrome. La cara de estos recién nacidos, con un cráneo abombado o una turricefalia y los ojos prominentes, sugieren el diagnóstico, pero estas características no son a menudo valoradas.4
Igualmente a menudo se observan malformaciones cardiovasculares y estenosis pulmonar, esta última generalmente con una válvula displásica, que constituye el trastorno característico de dicho síndrome.5 Los varones suelen tener criptorquidia y testículos pequeños (hipogonadales o no) y la pubertad puede llegar normalmente.2
La genética avanza, pero todavía no es convincente. La localización genética en el cromosoma 12 ha sido encontrada por varios especialistas en el estudio de este síndrome. 6 El trastorno resulta habitualmente esporádico, pero se han hallado antecedentes familiares de la enfermedad, generalmente en gemelos y con probabilidad monocigotos.7
Por lo infrecuente de la mencionada afección, los autores de este trabajo consideraron interesante describir el caso encontrado en nuestra área de salud, Centro Médico Universidad de las Ciencias Informáticas (UCI). En la literatura revisada solo se reporta un caso en edad pediátrica, localizado en el Hospital Orlando Pantoja, en el municipio Contramaestre, en la provincia de Santiago de Cuba.
Presentación del caso
Teniendo en cuenta que es en la APS donde se obtienen todos los datos relacionados con determinado proceso salud-enfermedad y su relación con la atención secundaria, expondremos el seguimiento del paciente hasta que fue visto por los autores de este trabajo en consulta durante el trabajo de dispensarización.
Paciente adolescente de 19 años, del sexo masculino y piel blanca, con antecedentes prenatales de ingreso hospitalario por polihidramnios. Nacido de un parto distócico (cesárea por desproporción cefalopélvica), para una edad gestacional de 40 semanas, que durante el proceso de dispensarización que se lleva a cabo en nuestra área de salud se encuentra que el paciente es portador del síndrome de Noonam.
Antecedentes patológicos personales:
- Año 1987: presentó linfangitis, bronconeumonía bibasal, neumonía de base izquierda, asma bronquial, se le dio seguimiento por alergia, y luego de realizárseles pruebas cutáneas que fueron positivas a la lana, la seda, el hongo, las plumas y el polvo, se le indicaron vacunas y el ketotifeno .
- Año 1989: presentó neumonía bibasal, asma, atelectasia base derecha, y se le diagnosticó el síndrome de Noonam. Padeció también de amigdalitis aguda, se le dio seguimiento por oftalmología por presentar coloboma de coroides y trastorno de refracción, y se mantiene usando cristales desde esa fecha.
- Año 1990: bronconeumonía bibasal. Tiene varios Rx de bronconeumonía, por lo que requirió tratamiento ambulatorio.
- A la edad de 4 años se interconsulta con cardiología por deformidad torácica para descartar afectación del área cardiaca, y no se le encuentran soplos u otras alteraciones.
- Recibe seguimiento por inmunología debido a trastornos de inmunidad celular transitoria adquirida, disminución de inmunoglobulina A (IgA) y alteración de la función opsono fagocitaria, por lo cual precisa de tratamiento con múltiples medicamentos (inferón, propóleo, factor de crecimiento y tramantadina). No se describe en la historia clínica individual del paciente la fecha de interconsulta con esta especialidad.
- A la consulta de genética acude por presentar alteraciones en la cara y el cuello. Se le indica cariotipo, que resultó normal, y electroforesis de proteínas. Al examen físico realizado se le encontró:
- Cabeza y cuello: implantación baja del pelo en la nuca, desviación mongoloide de los ojos, epicantus bilateral, orejas grandes algo despegadas (figura 1).
- Tórax: pectum excavatum. No se ascultan soplos, pero véanse otras características en la figura 2.
Es válido señalar que durante su estancia en nuestra universidad presentó episodios de catarro común, y requirió para su evolución la administración de antibióticos en una sola ocasión. No presentó otras enfermedades que pudieran desencadenar alguna descompensación en este paciente. Por otro lado, sus resultados docentes fueron evaluados satisfactoriamente
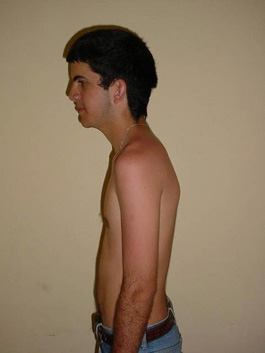
FIG. 1. Obsérvese la implantación baja del pelo en la nuca y pectum excavatum.
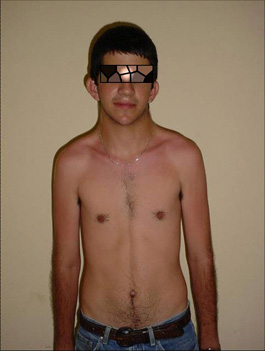
FIG. 2. Desviación mongoloide de los ojos, epicantus bilateral, orejas grandes algo despegadas.
Summary
Noonam's syndrome
The case of a 19-year-old male patient with Noonam's syndrome is described. It is characterized by chest deformity and minor malformations in the face. He was categorized at the familiy physician no. 1, corresponding to the Medical Center of the Computer Science University , in La Lisa municipality, Havana City. This patient is being followed up at present in the pneumology service of "Hermanos Ameijeiras" Clinical and Surgical Hospital .
Key words: Noonam's syndrome, adolescent, primary care, categorization.
REFERENCIAS BIBLIOGRÁFICAS
1. Disarz Algnaldo E, Ricardo Gómez A, Disarz Molinari E. Síndrome de Noonam. Rev Bras Méd. 1997;53(1-2):31-4.
2. Pacheco Alvarez LM, Sánchez Salcedo MA, Sánchez Pacheco DL, Rosales García J. Síndrome de Noonam. Presentación de un caso. MEDISAN. 2002; 6(1):86-90. Disponible en URL: http://bvs.sld.cu/revistas/san/vol6_1_02/san14102.htm
3. Nelson WE, Ricardo Gómez A, Disarz Molinari E. Síndrome de Noonam. En: Nelson WE, Vaughan VC, McKay RJ. Tratado de pediatría. 9na. ed. T2. La Habana: Editoral Científico-Técnica; 1998.p.1552.
4. Limal JM, Bonnet D, Le Bouc Y, Leheup B, Lyonnet S. Le syndrome de Noonam: une énigme. Arch Pédiatr. 1998;5:715-8.
5. Cruz Hernández M, Argemi J, Bueno M, Cardosa JJ. Tratado de pediatría. 5ta. ed. T1. Barcelona: Espaxs; 1983.p.676-941.
6. Ogata T, Muroya K, Tsukahara M. Noonan syndrome:genotype analysis of the Noonan syndrome critical region at chromosome 12 q in a three-generation family. Am J Med Genet. 1998;79:153-4.
7. Stein H, Hutton JJ, Koher PO, O´Rourke RA, Reynol HY, Samuels MA. Internal medicine. St Louis: Mosby Year Book; 1996:1286-9,1385.
Recibido: 15 de mayo de 2006. Aprobado: 14 de junio de 2006.
Dra. Emilia Yamilka Valdés Macola. Calle 238 # 3305, apartamento 1, bajos, entre 33 y 35, Reparto San Agustín, municipio La Lisa, Ciudad de La Habana, Cuba. E mail: evaldez@infomed.sld.cu
1Especialista de I Grado en Medicina General Integral.
2Licenciada en Enfermería.

