










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina General Integral
versión impresa ISSN 0864-2125
Rev Cubana Med Gen Integr vol.28 no.3 Ciudad de La Habana jul.-set. 2012
PÁGINA CULTURAL
Enfermedades raras
Rare deseases
MSc. Miguel Lugones Botell, Dra. Marieta Ramírez Bermúdez
Policlínico Universitario "26 de Julio". La Habana, Cuba.
INTRODUCCIÓN
Las enfermedades raras, minoritarias o huérfanas, como también suelen ser llamadas, incluidas las de origen genético, son aquellas por las que el enfermo puede encontrarse en peligro de muerte o de invalidez crónica y tienen una frecuencia (prevalencia) baja, de 5 casos por cada 10 000 habitantes en la comunidad,1 aunque existen otras clasificaciones.2
Hasta el momento se han identificado unas 7 000 enfermedades raras, en su mayoría crónicas y sin posibilidad de cura.2-6 Muchas de estas son causadas por cambios en los genes y se denominan enfermedades genéticas.2,3
Las enfermedades raras identificadas aumentan de manera progresiva, a pesar de que su número ya es muy elevado. Quienes las padecen tienen dolencias diferentes, pero carencias comunes.2 La lista de estas necesidades es muy amplia. Ya existe una estrategia con prioridades, entre las que destacan la creación de centros de referencia para diagnosticarlas, así como el deseo compartido por los afectados y sus familiares, de que se promocione la investigación aplicada, dirigida a hallar una solución para estas, de conjunto a la investigación básica que predomina en la actualidad.2
La primera vez que se habló de enfermedades raras fue en Estados Unidos en 1977,2 aunque su concepto se establece por primera vez en ese país a mediado de la década de los años 80 del pasado siglo7 y siempre estrechamente relacionado con el concepto de medicamentos huérfanos. Ambos términos se desarrollan en paralelo y ambos se dirigen a dar solución a los problemas que tienen las enfermedades de baja prevalencia;7 por los avances registrados, se ha asistido a un auténtico "boom" del censo de estas patologías. Además, de manera constante, se identifican nuevos genes implicados en su desarrollo.2
Las enfermedades raras son enfermedades con una alta tasa de mortalidad, pero de baja prevalencia, como anteriormente se dijo. Por lo general, comportan una evolución crónica muy severa, con múltiples deficiencias motoras, sensoriales y cognitivas; por lo tanto suelen presentar un alto nivel de complejidad clínica que dificultan su reconocimiento y diagnóstico.7 Entre 75 % y 80 % de estas dolencias infrecuentes, se debe a una alteración en los genes, mientras que entre 20 % y 25 % restantes, son enfermedades del sistema autoinmune, como la esclerodermia o el lupus eritematoso sistémico. El 3 % del ADN de todos los individuos está mutado, y todas las personas tienen entre 8 y 10 genes alterados, que podrían causar el desarrollo de una enfermedad rara.3
PROBLEMAS ASOCIADOS
La mayoría de las enfermedades afectan a más de un órgano vital, presentan un alto grado de complejidad diagnóstica, tienen un curso clínico crónico y son progresivamente debilitantes. Algunas otras son compatibles con una calidad de vida aceptable, siempre que se diagnostiquen a tiempo y se sigan adecuadamente. La esperanza de vida de todos estos pacientes está significativamente reducida.7
A menudo coexisten varias discapacidades, lo que acarrea múltiples consecuencias funcionales (la denominada multidiscapacidad o pluridiscapacidad). Estas discapacidades refuerzan la sensación de aislamiento y pueden ser una fuente de discriminación y reducir o destruir oportunidades educativas, profesionales y sociales. Por lo general, estamos hablando de personas dependientes de sus familias y con una calidad de vida reducida.7
La mayor parte de estas enfermedades son de herencia recesiva, es decir, la madre o el padre nunca los dos transmiten el gen causante, y por este motivo la mayoría de la población no las sufre. Hay pocas enfermedades de herencia dominante, en las que ambos progenitores transmiten los genes mutados. Esto explica que sean patologías de tan baja incidencia como para merecer el apelativo de "raras".3
¿CUÁNDO EMPLEAR EL APELATIVO "RARA"?
Para ser considerada rara, cada una de las enfermedades específicas solo puede afectar a un número limitado de personas, que en Europa está estipulado como menos de 1 por cada 2 000 ciudadanos, es decir, tiene baja frecuencia. Se estima que del 6 % al 8 % de la población mundial, aproximadamente, estaría afectada por una de estas patologías: 3 millones de españoles, 27 millones de europeos y 25 de ciudadanos estadounidenses.2
En España, unas 50 enfermedades raras afectan a algunos millares de personas, unas 500, a un centenar, y algunos millares solo a decenas de individuos. Un ejemplo de esto es que hay alrededor de 10 000 afectados por diversos tipos de anemia (talasemia, células falciformes...), cerca de 6 000 españoles tienen esclerosis lateral amiotrófica (ELA), no más de 5 000 sufren fibrosis quística, se conocen 5 000 casos de esclerodermia, unos 3 000 de miopatía de Duchenne y se han diagnosticado 2 500 casos de síndrome de Tourette, entre un listado muy amplio.2
Buena parte de estas enfermedades son crónicas y no tienen cura. En la actualidad, apenas se han desarrollado unos 50 medicamentos huérfanos nombre con el que se designa a los fármacos que se utilizan para tratarlas, número insignificante si se tiene en cuenta que hay unas 7 000 enfermedades.2 Ante esta falta de remedios farmacológicos, en ocasiones se trata a los pacientes con fármacos indicados para otras patologías, porque se ha demostrado que estos pueden mejorar su calidad de vida. Los médicos se los administran en los hospitales, en concepto de uso compasivo.
Sin embargo, el elevado costo de estos productos y el hecho de que sean medicamentos designados para otras patologías, limitan las opciones de tratamiento. Algunos enfermos no solo necesitan tratamientos farmacológicos, todavía sin diseñar, sino también otro tipo de cuidados.8
Los afectados por enfermedades dermatológicas como la epidermólisis bullosa una extrema fragilidad de la piel, que se ha comparado a la de una mariposa, y que sobre todo, afecta a niños necesitan cremas solares hidratantes, por motivos de salud, como si fueran fármacos.2
De la misma manera, en las enfermedades neurodegenerativas o neuromusculares, la rehabilitación mantenida a lo largo del tiempo, es una necesidad, porque mejora las capacidades residuales de los pacientes y permite su autonomía.9 Si el tratamiento rehabilitador se retrasa entre 4 o 5 años, los pacientes habrán recurrido ya, a una silla de ruedas.2
INVESTIGACIÓN APLICADA
Más investigación, pero aplicada. Esta es otra de las reclamaciones de los afectados, ya que la investigación que se realiza en la actualidad es básica, y busca los mecanismos que causan las enfermedades. También se ha planteado la necesidad de proveer de herramientas o protocolos a los médicos de familia, para ayudarles a detectar la posible existencia de una enfermedad rara.10 Los pacientes necesitan que se vaya más allá, y que la investigación se dirija a hallar una cura o a mejorar su calidad de vida. 2
Para conseguirlo, se debe lograr un "cambio de mentalidad de los investigadores, para que desarrollen fármacos contra estas dianas terapéuticas".2 En las originadas por un gen, como la ataxia de Friedreich, la solución curativa podría provenir de la sustitución del gen defectuoso (terapia génica), que se introduciría en el organismo a través de un agente vector. En otras enfermedades poligénicas o multifactoriales, hay que buscar otras soluciones terapéuticas, o al menos, algunas que mejoren la calidad de vida de los afectados.
La voluntad de algunos médicos que se han especializado en distintas enfermedades raras, ha favorecido la creación de centros de referencia.4 Se reclama, desde hace tiempo, la constitución de centros, servicios y unidades de referencia, donde los pacientes puedan someterse a todas las pruebas diagnósticas necesarias el mismo día, y recibir un diagnóstico lo antes posible. Deben ser centros que favorezcan la participación de los médicos en ensayos clínicos de su interés, garanticen el acceso a una atención multidisciplinaria y especializada de calidad, y ayuden a cualquier médico a conocer la ruta de actuación más adecuada para cada paciente diagnosticado. La intención es "aprovechar los recursos ya disponibles, y dar el reconocimiento que merecen a los profesionales que trabajan en estos centros".2
Hay diferentes empresas y laboratorios comprometidos en buscar y hallar soluciones para estos padecimientos. La importancia del deporte en el tratamiento de estas enfermedades, ha sido comprobado.9
Existe un registro de las 10 enfermedades más raras que se han encontrando mundialmente,11 las cuales resumimos a continuación:
1. Síndrome de hombre lobo (Hipertricosis lanuginosa congénita)
Frecuencia: Hay 40-50 casos documentados en todo el mundo. La incidencia natural (sin contar los casos en familias), se estima en 1 caso entre 1000 millones o 1 por 10 mil millones de habitantes.
Causa: Desconocida.
Descripción: Las personas que lo padecen están completamente cubiertas por un vello lanugo largo, excepto en las palmas de las manos y de los pies. La longitud a la cual puede llegar el vello, es de 25 centímetros.
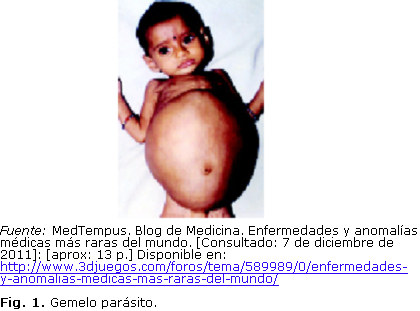
2. Gemelo parásito (Fetus in fetu)
Frecuencia: Alrededor de 100 casos documentados en todo el mundo.
Causa: Es una exageración del caso de los siameses. Dos gemelos no llegan a separarse completamente cuando son zigotos, y quedan unidos por alguna zona. Uno de estos gemelos crece sano, mientras que el otro se atrofia y queda en el interior del gemelo sano dependiendo completamente de él. Se desconoce por qué los gemelos no se separan correctamente.
Descripción: Cuando el feto hospedador consigue sobrevivir al parto, este puede mostrar un abombamiento en la zona donde se sitúe el feto parásito. El 80 % de las veces se encuentra en la región abdominal, pero también puede encontrarse en el cráneo, zona sacra, escroto (Fig. 1).
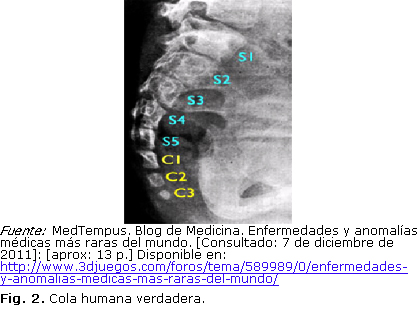
3. Cola humana verdadera (Cola vestigial)Frecuencia: Alrededor de 100 casos documentados en todo el mundo.
Causa: No se conoce en profundidad. Se cree que se produce por la mutación de los genes, encargados de producir la muerte celular programada de las células que estaban destinadas a formar una cola.
Descripción: Se observa la presencia de una cola vestigial en la zona final del sacro, a nivel cóccix. Esta cola está compuesta por tejido conectivo, músculos, vasos sanguíneos, nervios, piel, vértebras y cartílago (Fig. 2).
4. Progeria (Síndrome Hutchinson-Gilford)
Frecuencia: Alrededor de 100 casos documentados. Se estima que aparece un caso de progeria por cada 8 millones de nacimientos, aunque podría ser mayor, ya que muchas veces no llega a diagnosticarse.
Causa: La mayoría se producen por mutaciones de herencia autonómica dominante.
Descripción: Los individuos con progeria envejecen muy rápidamente desde la niñez. Al nacimiento tienen una apariencia totalmente normal, pero van creciendo cada vez más lentamente que los otros niños, y desarrollan una expresión facial muy característica. Pierden el pelo, adquieren arrugas, y padecen un daño severo de las arterias (aterosclerosis) que les lleva a la muerte, en los primeros años de la adolescencia (Fig. 3).

5. Síndrome de Proteus
Frecuencia: Actualmente hay documentados 200 casos en todo el mundo. Se estima que aparece 1 caso por más de 1 millón de nacimientos.
Causa: Desconocida.
Descripción: Existe una gran cantidad de malformaciones cutáneas y subcutáneas, con hiperpigmentación, malformaciones vasculares y crecimiento irregular de los huesos. Se produce el gigantismo parcial de los miembros, o el crecimiento excesivo de los dedos; mientras que algunas zonas del cuerpo crecen menos de lo que deberían. Todo esto provoca una desfiguración extrema de la persona, que la estigmatiza socialmente (Fig. 4). Josep Merrick, el famoso "hombre elefante", sufría de este síndrome.

6. Maldición de Ondina (Hipoventilación alveolar primaria)
Frecuencia: Entre 200-300 casos conocidos en todo el mundo. Por ser una causa de muerte súbita, se piensa que los casos conocidos son solo la punta del Iceberg, y que en realidad 1 bebé de cada 200 000 que nacen, podría tener esta enfermedad.
Causa: Parcialmente conocida. La principal causa es una mutación o varias del gen PHOX2B, de herencia autosómica dominante.
Descripción: En las formas más leves de la maldición de Ondina, el sujeto podrá seguir viviendo, pero debido a que el sueño no es reparador por la falta de oxígeno, durante el día estará somnoliento, se fatigará fácilmente, tendrá dolores de cabeza y aumento del nivel de glóbulos rojos.
Las formas más graves, en las que dormir significa una muerte segura, suelen aparecer desde el nacimiento, y la mayoría de los neonatos mueren, sin que muchas veces se llegue a saber la causa. Sin embargo, en aquellas personas en que la enfermedad ha empeorado progresivamente, y corre riesgo su vida cada vez que duermen, suele tratarse con ventilación asistida durante la noche. Aún así, a pesar de los tratamientos, cualquier descuido de quedarse dormido sin la oxigenoterapia indicada, significará la muerte.
7. Fibrodisplasia osificante progresiva
Frecuencia: Hay de 200-300 casos documentados en todo el mundo. Los pocos conocimientos que tienen los médicos acerca de esta enfermedad, propician que muchas veces no se diagnostique. Se estima que aparece 1 caso por cada 2 millones de nacimientos.
Causa: Es una enfermedad de herencia autosómica dominante. Se piensa que están implicados varios genes, encargados de sintetizar factores de crecimiento óseo.
Descripción: En esta enfermedad se dan episodios repetidos de inflamación de los tejidos blandos, se desarrollan tumores subcutáneos y en los músculos. Estas lesiones provocan la formación de hueso en sitios donde nunca debería producirse, como ligamentos, músculos, tendones, cápsulas articulares… Los traumatismos también desencadenan y hacen avanzar la osificación de los tejidos blandos (Fig. 5). Progresivamente el individuo irá perdiendo cada vez más movilidad, hasta que por imposibilidad de mover la musculatura encargada de la respiración (por estar osificada), muera por asfixia.

8. Hermafroditismo verdadero
Frecuencia: Existe alrededor de 500 casos documentados en todo el mundo. Se desconoce la frecuencia real en la población.
Causa: La persona hermafrodita es una quimera. Esta enfermedad se produce por la fusión de dos cigotos de distinto sexo. Es decir, primero un espermatozoide fecunda a un óvulo y después otro espermatozoide fecunda a otro óvulo más. Los cigotos que se forman y que estaban destinados a ser mellizos, se acaban fusionando y dan lugar a un único individuo, que genéticamente, es mujer y hombre al mismo tiempo. Se desconoce por qué se produce esta fusión de los cigotos.
Descripción: Los hermafroditas tienen tanto tejido ovárico como tejido testicular. Estos dos pueden encontrarse mezclados, lo que se llama ovotestis, o encontrarse por un lado un testículo, y por otro un ovario. Los genitales externos son ambiguos, y poseen componentes de ambos sexos. Las personas hermafroditas pueden tener apariencia femenina o masculina.
9. Síndrome de Moebius
Frecuencia: Hay alrededor de 80 casos documentados en España y 200 en Inglaterra. En Europa, aparecen al año aproximadamente 300 niños con este síndrome.
Causa: Desconocida. Ni siquiera se sabe si son los nervios, el tronco del encéfalo o los músculos los que están afectados en el origen de la enfermedad. Existen muchas y variadas hipótesis, pero sin pruebas que las respalden.
Descripción: Debido a que no se desarrollan algunos nervios faciales, las personas que nacen con este síndrome, carecen de expresión facial. No pueden sonreír, ni fruncir el ceño, etc. Tampoco pueden mover lateralmente los ojos, ni controlar el parpadeo. A menudo se les puede encontrar durmiendo con los ojos abiertos. Tienen grandes dificultades para succionar, tragar, hablar y para la realización de cualquier actividad en la que estén implicados los músculos de la cara.
10. Insensibilidad congénita al dolor
Frecuencia: Existen 100 casos documentados en Estados Unidos. Se desconoce la frecuencia en otras áreas, y muchas veces no se diagnostica porque pasa desapercibida.
Causa: Descubierta recientemente. Se debe a una mutación en un gen encargado de la síntesis de un tipo de canal de sodio, que se encuentra principalmente en neuronas encargadas de recibir y transmitir el estímulo doloroso.
Descripción: Son individuos totalmente normales en cuanto al tacto y la sensibilidad al frío, al calor, la presión y los cosquilleos. Sin embargo, ante cualquier acto que en personas normales provocaría dolor (como clavar una aguja), no hay ninguna sensación dolorosa. Como consecuencia de esto, suelen morir más jóvenes por traumatismos y lesiones varias, ya que no sienten ningún daño. Deben estar bajo supervisión en edades tempranas, para que no se lesionen ellos mismos.
ENFERMEDADES ULTRARARAS
Recientemente ha aparecido también la información de dos enfermedades ultrararas de gran importancia clínica para el pronóstico de las personas que las puedan padecer. Estamos hablando de:
La hemoglobinuria paroxística nocturna (HPN) y el síndrome hemolítico urémico atípico (SHUa) que consisten en la activación crónica e incontrolada del sistema del complemento con consecuencias graves y potencialmente mortales, "representan un reto diagnóstico, aunque por fortuna existen esperanzas terapéuticas en el momento actual."12
Se ha destacado la importancia de un diagnóstico precoz y de un tratamiento adecuado de estas enfermedades, para minimizar las graves consecuencias que estas dos entidades sistémicas y crónicas tienen en los pacientes. En el caso del SHUa, el diagnóstico "es muy importante porque la intervención rápida implica que no se produzca alteración sistémica y sobre todo que no se altere irreversiblemente la función renal, de modo que el enfermo no tenga que ser sometido a diálisis o a trasplante", en el caso de la HPN, se ha destacado que "la aplicación del tratamiento adecuado en pacientes con enfermedad muy sintomática, tiene un efecto muy favorable en su calidad de vida y en su pronóstico vital".12
La hemoglobinuria paroxística nocturna (HPN) es una enfermedad que afecta alrededor de 250 personas en España y que se caracteriza por una hemólisis crónica que puede producir graves daños a nivel sistémico como es la aparición de trombos, principal causa de muerte en estos pacientes.12
Por su parte, el SHUa es una enfermedad que aunque es crónica, se caracteriza por tener un proceso degenerativo muy rápido y que afecta tanto a adultos como a niños. A causa de esta afectación sistémica, más del 50 % de los pacientes fallecen, necesitan diálisis renal o presentan lesión renal permanente en el primer año desde el diagnóstico.
Tanto la HPN como el SHUa cursan con una sintomatología muy variada, como puede ser fatiga, afectación neurológica, episodios trombóticos, afectación renal, etc., por lo que resulta muy importante el papel del médico internista a la hora de diagnosticar este tipo de pacientes para lograr un tratamiento adecuado.12
Según se ha indicado eculizumab es un fármaco eficaz en ambas enfermedades, y esto se ha demostrado en ensayos clínicos. Por tanto hay una demostración científica de que corrige las principales consecuencias de estas dos enfermedades.
En el caso de la HPN, "disminuye tanto la cantidad de transfusiones que se realizan a estos enfermos, como el riesgo de que sufran una trombosis, de modo que los pacientes tratados con eculizumab tienen una supervivencia comparable a la de la población general"; mientras que en el SHUa "evita la aparición de microangiopatías trombóticas en órganos vitales, como por ejemplo el riñón, en una mayor proporción considerable de enfermos".12
Consideraciones finales
El tema de las enfermedades raras, resulta de gran interés como problema de salud, ya que aunque no constituyen padecimientos frecuentes, el hecho de que las padezcan grupos minoritarios de la población el ejemplo más elocuente lo constituye las enfermedades ultrararas, no le resta importancia, pues son personas que sufren y padecen afecciones para las cuales no existen tratamientos específicos. Por otra parte, los médicos deben tener conocimientos acerca de tales enfermedades, para poder pesquisar en la población, la posible presencia de estas.
REFERENCIAS BIBLIOGRÁFICAS
1. Categoría: Enfermedades raras. Wikipedia. [Consultado 29 de noviembre de 2011]: [aprox: 5 p.] Disponible en: http://es.wikipedia.org/wiki/Categor%C3%ADa:Enfermedades_raras
2. Consumer Eroski. Enfermedades raras. [Consultado 29 de noviembre de 2011]: [aprox: 3 p.]. Disponible en: http://www.consumer.es/web/es/salud/problemas_de_salud/2010/04/18 /192414.php
3. Enfermedades raras. [Consultado 6 de diciembre de 2011]: [aprox: 2 p.] Disponible en: http://www.nlm.nih.gov/medlineplus/spanish/rarediseases.html
4. Registro de enfermedades raras. [Consultado 6 de diciembre de 2011]: [aprox: 2 p.]. Disponible en: https://registroraras.isciii.es/Comun/Inicio.aspx
5. Enfermedades raras. [Consultado 6 de diciembre de 2011]: [aprox. 2 p.]. Disponible en: http://www.enfermedadesraras.es/
6. GeoSalud. Enfermedades raras. Tipos. [Consultado 6 de diciembre de 2011]: [aprox: 3 p.]. Disponible en: http://www.geosalud.com/enfermedades_raras/index.htm
7. Posada M, Martú-Arribas C, Ramírez A, Villaverde A, Abaitua I. Enfermedades raras. Concepto, epidemiología y situación actual en España. Anales del Sistema Sanitario de Navarra 2008 [Consultado 6 de diciembre de 2011];31(2): [aprox: 7 p.]. Disponible en: http://www.cfnavarra.es/salud/anales/textos/Vol31/sup2/suple2a.html
8. Pharmamar: Investigación biotecnológica. Tratamiento de enfermedades raras. [Consultado 29 de noviembre de 2011]: [aprox: 2 p.]. Disponible en: http://www.pharmamar.com/enfermedades-raras.aspx
9. FEDER. La importancia del deporte y sus beneficios en las enfermedades raras. [Consultado 29 de noviembre de 2011]: [aprox: 2 p.]. Disponible en: http://www.enfermedades-raras.org/index.php?option=com_content&view=article&id=1355:la-importancia-del-deporte-y-su-beneficio-en-las-enfermedades-raras&catid=1:latest-news
10. Medicina y Humanidades. Noticias. Pacientes y médicos crean una plataforma para la investigación de enfermedades raras. [Consultado 19 de diciembre de 2011]: [aprox: 4 p.]. Disponible en: http://www.jano.es/jano/actualidad/ultimas/noticias/janoes/pacientes /medicos/crean/plataforma/investigacion/enfermedades/raras/_f-11+iditem-15795+idtabla-1
11. MedTempus. Blog de Medicina. Enfermedades y anomalías médicas más raras del mundo. [Consultado 7 de diciembre de 2011]: [aprox: 13 p.]. Disponible en: http://www.3djuegos.com/foros/tema/589989/0/enfermedades-y-anomalias-medicas-mas-raras-del -mundo/
12. Jano y Humanidades. Jano.es. El bloqueo del sistema del complemento comporta beneficios en dos enfermedades ultra-raras. [Consultado: 4 de junio de 2012]: [aprox. 2 p.]. Disponible en: http://www.jano.es/jano/actualidad/ultimas/noticias/janoes/bloqueo/sistema /complemento/comporta/beneficios/dos/enfermedades/ultra-raras/_f-11+iditem -17230+idtabla-1
Recibido: 14 de marzo de 2012.
Aprobado: 16 de abril de 2012.
Miguel Lugones Botell. Policlínico Universitario "26 de Julio". Calle 72 entre 13 y 15, municipio Playa. La Habana, Cuba. Correo electrónico: lugones@infomed.sld.cu
