










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La poroqueratosis (PQ) fue descrita por el dermatólogo italiano Vittorio Mibelli en el 1893, al considerar que se originaba en el "poro" sudoríparo, pero con los nuevos estudios se describe como una dermatosis que se caracteriza por un trastorno de la queratinización.1,2,3 Su incidencia es baja a nivel mundial, se reportan alrededor de 250 casos.2 Puede afectar cualquier raza y sexo, con predominio en niños y adultos jóvenes.3
Se plantea que es una genodermatosis con un patrón de herencia autosómico dominante con penetrancia variable, aunque se han visto algunos casos que pueden ser a consecuencia de mutaciones espontáneas. Además, cuando en un mismo individuo o en varios miembros de una misma familia coinciden varias formas clínicas, sugiere que puede tratarse de diferentes expresiones fenotípicas de un mismo trastorno genético.2,4) En una persona con predisposición genética, se presume que las manifestaciones clínicas son desencadenadas por factores agregados, tales como la radiación ultravioleta, ya sea de origen natural o artificial. Asimismo, son desencadenantes de la enfermedad diversas causas de inmunosupresión como: infección por el virus del HIV, insuficiencia hepática o renal crónicas, trasplante de órganos y quimioterapia. Otros factores mencionados son: hemopatías, neoplasias, diuréticos tiazídicos, exposición a rayos X e incluso a radiación con electrón beam.3,4,5
En la literatura médica se ha reportado que en todas las formas de PQ se puede presentar la degeneración maligna de las lesiones, con un riesgo de 7,5 % a 11 %.6) Además, se plantea la hipótesis que la inestabilidad cromosómica y la reducida vigilancia inmune con sobreexpresión de la proteína p53 pueden ser la causa para el desarrollo de enfermedades malignas cutáneas.2,6
Haber identificado en la consulta de dermatología en conjunto con la de genética comunitaria del municipio de Manatí varios pacientes en una misma familia con esta enfermedad, nos motivó a realizar este trabajo, con el objetivo de evaluar el riesgo genético, para poder realizar la profilaxis y control de los miembros afectados y su descendencia.
PRESENTACIÓN DEL CASO
Se trata de una paciente femenina de 38 años de edad, procedencia urbana y piel blanca. Se desempeña como profesora de secundaria básica. Esta refiere antecedentes patológicos personales de asma bronquial sin crisis frecuentes. No ha presentado alergias a medicamentos. No ha tenido contacto con sustancias tóxicas, ni posee hábitos tóxicos.
Acude a la consulta de dermatología porque desde los 24 años de edad presenta lesiones a nivel de miembros superiores e inferiores con oscurecimiento de la piel y prurito. Refiere que en la familia existen varios miembros que presentan lesiones similares como su abuela materna, madre, tíos y una prima.
Examen físico
Paciente sin afectación del estado general.
Mucosas: húmedas y normocoloreadas.
Tejido celular subcutáneo: no presenta edema
Aparato respiratorio: murmullo vesicular normal, no presenta estertores. FR 18x´
Aparato cardiovascular: ruidos cardiacos rítmicos, no presenta soplo. T/A: 110/70, FC75x´
Abdomen: sigue los movimientos respiratorios, blando a la palpación, depresible, no presenta viceromegalia. RHA presentes.
SNC: conciente, orientada en tiempo espacio y persona. Reflejos y sensibilidad normales.
Se identificaron lesiones en la piel en forma de placas anulares, miembros superiores e inferiores, de tamaño y forma irregular. Se encontraron, además, otras lesiones atróficas sin pelo y anhidróticas. Algunas de estas últimas hiperpigmentadas, con un borde ligeramente hipertrófico que las separa de la piel normal (figuras1 y 2).


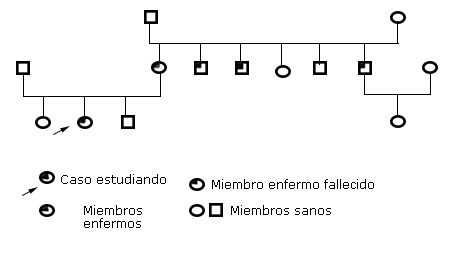
Se valoró el caso por genética. Se confirmó que la abuela de 76 años presenta lesiones con similares características, a su vez refirió que 4 de sus 6 hijos también las presentaron y otra nieta de 20 años. Esto nos permitió pensar en una genodermatosis con carácter dominante, y de estas, la Poroqueratosis de Mibelli (Fig. 3).
Se le indicaron varios exámenes complementarios, con los siguientes resultados:
Hemoglobina: 12,3 g/l
Hematocrito: 0,38
Glicemia: 4,2mmol/l
Creatinina: 56mmol/l
Urea: 2,3 mmol/l
Colesterol: 3,18 mmol/l
Trigicéridos: 1,1 mmol/l.
Ultrasonido abdominal: hígado que no rebasaba el reborde costal; vesícula, páncreas, riñones y bazo normales.
Rx de tórax: sin alteraciones.
Fondo de ojo: sin alteraciones.
La biopsia de piel resultó el análisis más significativo. La muestra para el análisis se tomó de la lesión que más le afectaba a la paciente, el resultado histopatológico confirmó la presencia de PQ.
Se le orientó seguimiento por dermatología, con tratamiento medicamentoso citostático tópico (5-fuoracilo) y una evolución favorable. En el caso de genética, el objetivo consistió en la asesoría genética a la familia a partir de charlas sobre la enfermedad y la posibilidad de aparecer en la descendencia al tener en cuenta el patrón de herencia.
DISCUSIÓN
La PQ es una enfermedad poco frecuente a nivel mundial y rara en nuestro país,7 se reportan casos con cierta frecuencia en algunos países como Estados Unidos de América, se manifiesta en personas rubias, afecta los hombres en proporción 2:1 frente a las mujeres y se desarrolla con frecuencia en la niñez.6,7 En un estudio realizado en la provincia de Cienfuegos, se identificó la enfermedad en varones trigueños, quienes debutaron durante la infancia.6 En este estudio predominaron las mujeres, de piel blanca que debutaron después de los 20 años.
Entre las manifestaciones clínicas de la PQ, se describe que las lesiones pueden localizarse en cualquier sitio del tegumento: cara, extremidades, genitales, glúteos y otras partes del cuerpo. Una generalidad es que permanecen asintomáticas, a veces con prurito discreto durante algunos períodos del año, crecen con rapidez en sitios afectados por traumas o quemaduras. Pueden ser hiper o hipopigmentadas con descamación discreta, de centro atrófico, sin pelo, con marcada anhidrosis y con tamaños que varían desde milímetros a varios centímetros.7,8 El caso estudiado presentó lesiones con las características descritas en los miembros superiores e inferiores, refirió prurito y sin ningún antecedente de traumas o quemaduras.
Es una enfermedad considerada de causa genética con un patrón de herencia autosómico dominante debido a la presencia de clones celulares que muestran grados variables de displasia, se ha demostrado inestabilidad en el brazo corto del cromosoma 3, aunque la causa no se conoce.9) En la valoración genética del caso se puso de manifiesto el patrón de herencia antes mencionado, teniendo en cuenta el árbol genealógico que se realizó durante la investigación.
La confirmación de la sospecha clínica se realiza con la histopatología, se destaca que un exhaustivo examen físico y conocimiento de la semiología deviene prediagnóstico. La administración tópica de iodopovidona sobre una lesión sospechosa permite distinguir los bordes de la lesión correspondiente debido a su coloración más intensa. Se facilita así el diagnóstico en casos poco evidentes.9
Thomas y otros10 describieron un método no invasivo y expedito para identificar el borde queratótico de la lesión, compatible histológicamente con la laminilla cornoide. Para ello se utiliza la violeta de genciana. La dermatoscopía mejora la visualización del borde hiperqueratósico y permite diferenciarlo de lesiones clínicamente similares como: queratosis actínica, enfermedad de Bowen y otras. Otro grupo de dermatólogos italianos describen las lesiones amarronadas hacia la periferia de la misma, con un área central de aspecto cicatrizal (formada) y escasas estructuras globulares de color rojo. Delfino,10 utiliza un microscopio estereoscópico para observar algunas de las lesiones diez veces magnificadas y lo compara con la observación de un cráter volcánico desde la altura. .En nuestro estudio no fue necesario aplicar estas técnicas, ya que los antecedentes familiares, la clínica y el estudio anatomopatológico realizado confirmaron la enfermedad.
El tratamiento de la PQ debe ser individualizado. Esto se basa en el tamaño de la lesión, en la localización anatómica, en consideraciones estéticas y en la preferencia del paciente.9,10 La protección del sol, el uso de emolientes y la observación de signos de malignidad deben estar presentes. En el caso descrito se pudo ver la mejoría de las lesiones tras el uso tópico del medicamento y las medidas orientadas.
En su diagnóstico diferencial se integra el grupo de las manifestaciones dermatológicas de los procesos internos, incluidos los fenómenos paraneoplásicos.11 Su manejo es esencial, debido a que requiere atención multidisciplinaria dado el hecho de que los individuos de varias generaciones pueden ser afectados. En tal sentido, es relevante el conocimiento de los miembros del riesgo genético para evitar los factores desencadenantes y la aparición de la enfermedad.

