










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
La inmunodeficiencia común variable (IDCV) pertenece a los errores innatos de la inmunidad (EII) o inmunodeficiencias primarias, clasificando como una deficiencia predominantemente de anticuerpos. La IDCV cursa con niveles bajos de inmunoglobulinas (IgG, IgM y/o IgA) y se caracteriza por una capacidad disminuida en la producción de anticuerpos específicos, derivando en infecciones recurrentes de predominio sino-pulmonar y en muchos casos complicaciones no infecciosas (autoinmunidad, bronquiectasias, enfermedad pulmonar crónica, granulomas y malignidad) en sujetos en los que se descartaron otras causas de hipogammaglobulinemia.1,2) A pesar de tratarse de un trastorno genético, la instauración de las manifestaciones clínicas cardinales tiene su pico en la edad adulta,3) haciendo de su diagnóstico un reto para el médico general y especialista. A continuación, presentamos el caso de un hombre de 35 años con infecciones sino-pulmonares a repetición e hipogammaglobulinemia grave, en quien se descartaron las causas usuales de inmunosupresión, se llegó al diagnóstico definitivo de IDCV, se resalta la importancia de pensar en los EII en pacientes adultos.
Presentación del caso
Hombre de 35 años que se presenta a la consulta de Medicina Interna - Inmunología refiriendo un cuadro clínico de 3 años de evolución consistente en múltiples episodios de infecciones sino-pulmonares en los últimos meses, presentaba tos productiva, dificultad respiratoria y pérdida de peso no intencional de aproximadamente 8 kg.
El primer episodio de neumonía (no complicada) ocurrió a los 32 años, fue manejado con terapia antibiótica empírica con ampicilina-sulbactam durante 7 días, con posterior resolución completa. A los 34 años presentó un segundo cuadro de neumonía, esta vez complicada por un derrame pleural izquierdo de características exudativas, la tomografía axial computarizada (TAC) de tórax muestra una consolidación en el lóbulo inferior izquierdo asociado a derrame pleural cercano al 30 % además con presencia de bronquiectasias, patrón de árbol en gemación e infiltrados en vidrio esmerilado (Fig. 1B), la fibrobroncoscopia mostró endobronquitis, los estudios del lavado bronco alveolar y líquido pleural descartaron infección por M. tuberculosis (GenXpert, baciloscopias con Auramina, adenosina deaminasa y cultivo, todos negativos). La histopatología de las biopsias endobronquiales mostró inflamación crónica, sin granulomas ni microorganismos detectados por coloraciones especiales. Las pruebas de ELISA para VIH en 3 oportunidades fueron negativas. Requirió manejo hospitalizado por 13 días, siendo tratado inicialmente con Claritromicina + Cefepime + Vancomicina, esta última reemplazada por Linezolid dada evolución clínica estacionaria. Los hemocultivos fueron negativos, el paciente mejoró significativamente y se le dio egreso con estudios ambulatorios.
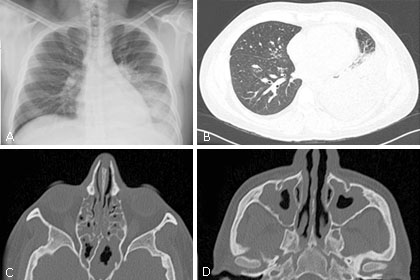
El paciente, 5 meses posterior al egreso, se complica con una sinusitis aguda con importante respuesta inflamatoria sistémica, el TAC de senos paranasales evidenció ocupación y engrosamiento mucoso compatible con sinusitis crónica agudizada. Otorrinolaringología realizó etmoidotomía endoscópica y toma de cultivos previo al inicio de terapia antibiótica empírica, sin aislamiento microbiológico, pero con mejoría clínica significativa.
En controles ambulatorios, un nuevo TAC de tórax evidenció disminución del patrón de árbol en gemación y este se consideró secundario a broncoaspiración crónica por enfermedad de reflujo gastroesofágico (ERGE) por una impedanciometría positiva para reflujo ácido, diurno y nocturno, sumado a sinusopatía crónica (ya intervenida), sin embargo, se realiza una segunda fibrobroncoscopia y se descarta infección por M. tuberculosis y hongos, con cultivo positivo para H. influenzae multisensible. Las biopsias endobronquiales no tenían cambios respecto a la inicial y la citología era normal. Se instaura tratamiento con quinolonas durante 8 días y contra ERGE y se continuaron estudios de forma ambulatoria en búsqueda de etiología reumatológica, solicitando perfil de autoanticuerpos, ANAS, ENAS, p-ANCAS, c-ANCAS, complemento sérico C3, C4 y VSG, los cuales resultan dentro de rangos de normalidad, así como hemogramas sin evidencia de alteraciones cuantitativas en líneas celulares (Tabla 1).
Tabla 1 Laboratorios de evaluación inmunológica y virológica
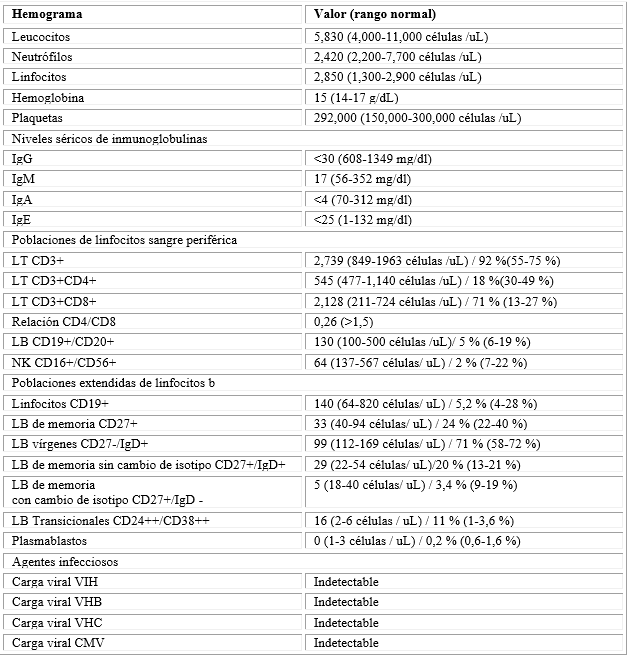
*Poblaciones totales y/o proporciones alteradas resaltadas en negrilla.
El paciente persistió sintomático (predominantemente respiratorio superior) y a los 35 años de edad desarrolla una pansinusitis bilateral (Fig. 1C-D), fue intervenido otra vez por parte de ORL con etmoidectomía anterior y posterior, antrostomía bilateral de esfenoides vía endoscópica más lavado con Anfotericina B. Nuevamente, los estudios microbiológicos de muestras quirúrgicas resultan negativos y el reporte de biopsia evidencia inflamación aguda y crónica con pólipos inflamatorios y eosinófilos escasos.
Hasta ese momento el paciente contaba con dos episodios de neumonía de aparente origen bacteriano, uno de ellos complicado por derrame pleural, así como dos episodios de sinusitis agudizada que demandaron intervención quirúrgica e incluso lavado intracavitario con antifúngico. En el desarrollo de su evolución clínica se había descartado tuberculosis pulmonar, infección por VIH, neoplasia pulmonar y patología autoinmune.
En la consulta de inmunología se indagaron sobre antecedentes personales adicionales, negó consanguinidad parental, consumo de sustancias psicoactivas, cigarrillo o exposición a tóxicos, sin embargo, el paciente refería automedicación con antibióticos (azitromicina o amoxicilina/clavulonato). Negó muertes tempranas o no explicadas en familiares, con único antecedente de cáncer pancreático en familiar de línea paterna. El paciente era enfático en que era previamente sano. Al examen físico el paciente no evidenciaba signos de dificultad respiratoria, los signos vitales se encontraban en parámetros normales, sin embargo, lucía crónicamente enfermo y adelgazado.
Se consideró que el paciente cursaba con un patrón de infección recurrente anormal, no asociado a inmunosupresión infecciosa, neoplásica o farmacológica, se propuso como impresión diagnóstica una inmunodeficiencia primaria de instauración tardía, por lo cual se realizó una evaluación de la respuesta inmune humoral y celular. Los resultados mostraron hipogammaglobulinemia en rangos de agamaglobulinemia para la IgG, IgM e IgA, las poblaciones linfocitarias T (LT) sin linfopenia, pero con inversión de la relación CD4+:CD8+ (0.26), una cantidad normal de linfocitos B (LB) y evidencia de linfopenia NK (NK). Las poblaciones extendidas de LB evidenciaron disminución en los LB de memoria con cambio de isotipo y en los plasmablastos, con un exceso en la proporción de LB transicionales, confirmando una capacidad disminuida para generar células productoras de anticuerpos efectoras y de memoria (Fig. 2D).
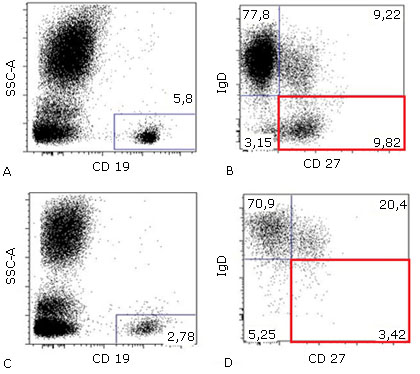
Fig. 2 Diagramas de puntos (dot plot) de citometría de flujo. A) Cuantificación normal de LB totales sobre la población de leucocitos en un individuo sano. B) Cuantificación normal de LB de memoria sin cambio de isotipo (CD27+/IgD +) y LB de memoria con cambio de isotipo (CD27+/IgD-) en un individuo sano. C) Cuantificación normal, aunque con tendencia baja de LB totales sobre la población de leucocitos del paciente mencionado. D) Citometría de flujo con proporción anormal de linfocitos B de memoria sin cambio de isotipo (CD27+/IgD +) y linfocitos B de memoria con cambio de isotipo (CD27+/IgD -) en el paciente mencionado. Nótese la marcada diferencia en la población de LB de memoria con cambio de isotipo (productores de IgG, IgM, IgE) entre un paciente sano y uno afectado por IDCV (cuadrados rojos)
Con todos los datos anteriores se configuró el diagnóstico de IDCV.4 Se descartaron infecciones crónicas (VIH, VHC, VHB, CMV) por medio de cargas virales, teniendo en cuenta que las pruebas serológicas podrían resultar falsas negativas dada su incapacidad para producir anticuerpos. Adicionalmente, se realizó Exoma clínico (Next Generation Sequencing) buscando mutaciones en genes relacionados con la respuesta inmune, sin embargo, no se identificaron variables patogénicas.
El tratamiento con inmunoglobulina humana en sustitución es una indicación absoluta en este caso, razón por la cual se inició a una dosis de 800 mg/kg intravenosa cada 4 semanas, acorde con los lineamientos colombianos en pacientes con inmunodeficiencias primarias con daño pulmonar estructural.5 Ahora cuenta con dos años de terapia de reemplazo, hace aproximadamente un año se realizó el cambio por inmunoglobulina humana subcutánea facilitada con hialuronidasa por decisión consensuada con el paciente, manteniendo niveles valle de inmunoglobulina sérica dentro de los rangos fisiológicos esperados y evolucionando clínicamente sin nuevos episodios infecciosos.
Discusión
Los EII son un grupo heterogéneo de patologías consideradas como enfermedades raras o huérfanas, pero en conjunto pueden alcanzar prevalencias de hasta 1/2.000 personas.6 Se caracterizan por ser defectos primarios de la respuesta inmunológica mediados por trastornos monogénicos o poligénicos y expresa fenotipos clínicos muy diversos e inespecíficos, factores determinantes en su retraso diagnóstico.
La IDCV es el EII clínicamente significativo más frecuente de todos,3) el cuadro clínico “variable” puede llevar al paciente a consultar a diferentes especialidades médicas lo que favorece un retraso diagnóstico que en promedio es de 7 años, en algunos casos llega hasta los 15 años. Los afectados se pueden clasificar en dos principales fenotipos clínicos, “no complicados” o “complicados”, los primeros sufren manifestaciones predominantemente infecciosas (30 % de pacientes) y los segundos presentan adicionalmente manifestaciones clínicas inflamatorias (enfermedades granulomatosas, linfadenopatías), autoinmunidad (principalmente citopenias) y malignidad (70 % restante de los pacientes), además, es preocupante el compromiso grave de la sobrevida en los casos complicados, que reduce la expectativa de vida hasta en 30 años.7,8
El caso presentado tuvo múltiples oportunidades de atención en servicios de atención primaria, urgencias y hospitalización con equipos multidisciplinarios de especialistas a lo largo de 3 años, durante los cuales se otorgó el estatus de “inmunocompetente” basados principalmente en la ausencia de infección por VIH o autoinmunidad, pero sin una evaluación objetiva de los componentes de la respuesta inmune susceptibles de ser valorados en la práctica clínica, como son los niveles séricos de inmunoglobulinas, las poblaciones linfocitarias (LT, LB y NK), prueba de estallido respiratorio de fagocitos, entre otros,9) que de haberse considerado podrían haber contribuido a un diagnóstico y tratamiento más temprano. Se destaca un primer mensaje para el clínico: para aproximarse a afirmar un estatus de inmunocompetencia se deben agotar múltiples recursos diagnósticos, con el objetivo de descartar causas de inmunosupresión no solamente infecciosas, autoinmunes, neoplásicas, metabólicas o tóxicas, si no también inmunológicas primarias.
Fenotípicamente podemos clasificar al paciente dentro del grupo de pacientes “complicados” por la presencia de bronquiectasias, las cuales se interpretaron y atribuyeron inicialmente como una complicación de la sinusopatía crónica y la enfermedad por reflujo gastroesofágico presente en el paciente, enmascarando el hecho de que la sinusopatía y las neumonías recurrentes eran una consecuencia de un estado de susceptibilidad inmunológica no identificado. Esto deja un segundo mensaje clave para el clínico: la identificación de un patrón anormal de infección, entendiéndose cómo una alta recurrencia de infecciones, infecciones causadas por microorganismos oportunistas, infecciones que no respondan a tratamiento estándar, que sean prolongadas o que lleven a cuadros graves de manejo en cuidado intensivo y/o cuadros clínicos con manifestaciones inflamatorias o autoinmunes de origen incierto, ameritan una evaluación básica del sistema inmune en búsqueda de trastornos inmunológicos primarios10) y redireccionamiento a un especialista en inmunología.
Dentro del perfil de evaluación inmunológica, además de la evidencia de hipogammaglobulinemia, hallazgo cardinal en esta patología, cabe resaltar la presencia de una relación inversa a la esperada entre los LTCD4+ y los LTCD8+, dada por una linfocitosis de LTCD8+, dato que sugiere una alta expansión crónica de esta población celular comparada con los LTCD4+, así como una linfopenia de NK, ambos hallazgos también descritos en pacientes con IDCV de fenotipo complicado.11,12,13
La aproximación a un diagnóstico genético utilizando un exoma con tecnología de secuenciación de nueva generación no arrojó variantes genéticas que se asociaran o explicaran la IDCV en el paciente, hallazgo que se correlaciona con lo descrito en la literatura, donde las mutaciones causantes de enfermedad solo se identifican 20 % de los afectados por esta enfermedad,14) plantea que la IDCV corresponde a una enfermedad donde diferentes mecanismos genéticos coexisten para arribar a una misma expresión clínica o donde múltiples causas monogénicas, poligénicas o epigenéticas aún se desconocen.15 Sin embargo, también se ha demostrado que la búsqueda de una causa genética es de alto valor clínico e investigativo, bien sea para descartar otras causas de inmunodeficiencia primaria, como para identificar variantes que pudieran tener significado clínico en pacientes con fenotipos complicados, aportando nuevos datos para la disección de una enfermedad en continua caracterización,16,17 lo que justifica la realización de un exoma en este caso.
La meta del tratamiento es el reemplazo de las inmunoglobulinas, administrando inmunoglobulina humana de forma intravenosa (IGIV) mensualmente o subcutánea (IGSc) semanalmente, para disminuir la tasa de infección y otras complicaciones inflamatorias. En Colombia existen diferentes formas de inmunoglobulina humana autorizadas para tratamiento de sustitución en inmunodeficiencias por el INVIMA, la cual debe administrarse a dosis entre 400-800 mg/kg depende de los niveles valle y la respuesta clínica. También se dispone de inmunoglobulina humana subcutánea para aplicación semanal a dosis de 100- 200 mg / kg y recientemente ingreso al país una presentación de inmunoglobulina humana subcutánea (10 %) facilitada con hialuronidasa.5 En aquellos casos que se presenten con niveles bajos de linfocitos T CD4+ (<200celuls/uL) se recomienda el uso de profilaxis antibiótica con trimetoprim/sulfametoxazol.3
Los errores innatos de la inmunidad en adultos deben ser una posibilidad diagnóstica para el médico general y especialista. En nuestro medio contamos con la experticia, tecnología y metodologías para confirmar su diagnóstico. Este reporte de caso pretende llamar la atención sobre una patología que requiere un alto índice de sospecha que sumado a su baja prevalencia tiene un impacto deletéreo en el pronóstico de los pacientes.

