










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Ortopedia y Traumatología
versão On-line ISSN 1561-3100
Rev Cubana Ortop Traumatol v.15 n.1-2 Ciudad de la Habana jan.-dez. 2001
Hospital Pediátrico Docente
“Eliseo Noel Caamaño”. Matanzas
Hipofosfatasia. Presentación de un caso
Dra. Elsa Luna Ceballo,1 Dra. María E. Domínguez Pérez2 y Dra. Rudbeckia Álvarez Núñez3
Luna Ceballo E, Domínguez Pérez ME, Álvarez Núñez R. Hipofosfatasia. Presentación de un caso. Rev Cubana Ortop Traumatol 2001;15(1-2):87-9
Resumen
Se presenta un paciente con hipofosfatasia, trastorno hereditario del metabolismo óseo de rara ocurrencia. Hacemos referencia a su clasificación y forma de herencia. Describimos las características clínico-radiológicas y los exámenes de laboratorio clínico que nos permitieron realizar el diagnóstico en este caso.
DeCS: HIPOFOSFATASIA/genética; HIPOFOSFATASIA/metabolismo; RADIOGRAFIA/métodos; FOSFATASA ALCALINA.
La hipofosfatasia es un trastorno hereditario del metabolismo de gravedad variable, fue descrito, por primera vez, en 1948 por Rathbun.1 Los tres criterios fundamentales para el diagnóstico de la misma son: una disminución de la actividad de la fosfatasa alacalina en el suero y en muchos tejidos, una osificación irregular e incompleta del cartílago y del hueso en crecimiento, visible en las radiografías y en los cortes histológicos, y una mayor excreción urinaria de fosforiletanolamina.1,2
Existen varios tipos de hipofosfatasias con fenotipos superpuestos, pero con distintas formas de herencia, por lo que se postula una transmisión autosómica recesiva o dominante.3 Esta entidad de rara ocurrencia se presenta con una frecuencia de 1 por cada 100 000 nacimientos.4
En la literatura revisada encontramos diferencias entre los autores al clasificar las formas de presentación.3,5,6 Básicamente se conocen tres o cuatro variedades aunque no siempre es posible diferenciarlas entre sí.5
- Forma Neonatal: Se presenta después del nacimiento. Los niños más gravemente afectados nacen muertos o fallecen poco después. Llaman la atención por ser pequeños, con extremidades cortas y arqueadas, articulaciones engrosadas y huesos craneales blandos. En el transcurso de unos días presentan irritabilidad, dificultad respiratoria, fiebre, etc.
- Forma infantil: Después de un período asintomático van apareciendo gradualmente los mismos signos clínicos. La mayoría sobreviven y más adelante experimentan una mejoría. Las secuelas tardías consisten en corta estatura, piernas arqueadas, retraso de la marcha, craneosinostosis, pérdida prematura de los dientes y caries.7-10
- Forma de la infancia: Se presenta entre los seis meses y los dos años e incluso más tarde, se caracteriza por manifestaciones moderadas de la enfermedad, semejante a la forma infantil. El cráneo no suele mostrar craneosinostosis.
- Forma adulta: Caracterizada fundamentalmente por desmineralización ósea y fracturas. A menudo existe una historia familiar de hipofosfatasia.
Los hallazgos radiológicos son característicos de la forma neonatal, observándose una rarefacción esquelética generalizada, huesos cortos y arqueados, metáfisis ensanchadas e irregularmente clasificadas, fracturas y suturas craneales que parecen ser amplias a causa de la deficiente osificación. En las otras formas los hallazgos radiológicos pueden ser sutiles y no tan característicos.5,11
La escasa frecuencia con que se presenta esta enfermedad y el pronóstico reservado de muchos de estos casos, la convierte en una entidad de gran interés médico, de ahí que sea necesario realizar un rápido diagnóstico para aliviar los trastornos del paciente.
Presentación del caso
Paciente prematura, sexo femenino, procedencia rural, de 19 días de edad, nacida de parto extrahospitalario, ingresada en el Hospital Pediátrico por presentar dificultad respiratoria, polipnea, secreción nasal serosa y fiebre de 38°C. Al examen físico se encontró baja talla (menor del tercer percentil), extremidades cortas y arqueadas con engrosamiento de articulaciones fundamentalmente muñecas, tobillos y rodillas, cráneo blando, frente amplia, nariz en silla de montar, nares antevertidos, caja torácica estrecha e hipotonía generalizada.
Los exámenes de laboratorio clínico arrojaron los siguientes resultados: Hb 11 g/L; fosfatasa alcalina en suero: disminuida; calcio en sangre: aumentado; fósforo en sangre: normal; eritrosedimentación: acelerada.
En los estudios radiológicos encontramos una rarefacción ósea generalizada, caja torácica estrecha, costillas cortas con expansión y concavidad de sus extremidades anteriores, arqueamiento de huesos largos, ensanchamiento y deformidad en copa de las metáfisis con mineralización irregular de las mismas, centros de osificación de contornos irregulares y pobremente clasificados (figura). La radiografía de cráneo muestra una disminución de la mineralización ósea con suturas y fontanelas amplias.
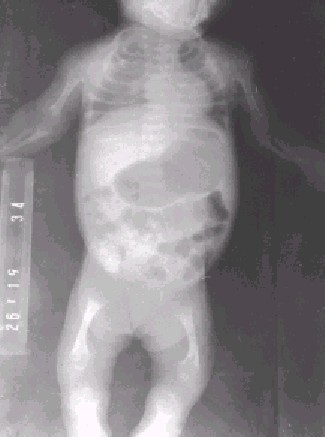
FIG. Obsérvese la rarefacción ósea y el resto de los signos radiológicos señalados en el texto.
Discusión
El examen físico de esta paciente, asociado a los exámenes de laboratorio, demuestra la característica disminución de la actividad de la fosfatasa alcalina y los típicos hallazgos radiológicos los que son compatibles con el diagnóstico de una hipofosfatasia.
Los pacientes que tienen el tipo más severo de este trastorno, generalmente, mueren en los primeros seis meses de vida, debido a la insuficiencia respiratoria. En el caso presentado, la temprana edad de comienzo de la enfermedad, asociado a la estrechez de la caja torácica y los síntomas de dificultad respiratoria de rápido debut hacen que el pronóstico sea sombrío.
La similitud de la hipofosfatasia con el raquitismo desde el punto de vista clínico y de algunos aspectos radiológicos, nos lleva a hacer un estudio exhaustivo del paciente con vista a identificar los criterios fundamentales: clínicos, radiológicos y de laboratorio que nos permitan establecer un diagnóstico definitivo y precoz.
Summary
A patient with hypophosphatasia, a rare hereditary disorder of the bone metabolism is presented. Reference is made to its classification and inheritance form. The clinical and radiological characteristics and the clinical laboratory tests that allowed us to make the diagnosis in this case are also described.
Subject headings: HYPOPHOSPHATASIA/genetics; HYPOPHOSPHATASIA/metabolisms; RADIOGRAPHY/methods; ALKALINE PHOSPHATASE.
Résumé
Un patient atteint d‘hypophosphatasie, un rare trouble héréditaire du métabolisme osseux, est présenté. On fait référence à sa classification et forme d‘hérédité. Les caractéristiques clinico–radiologiques et les épreuves de laboratoire clinique nous permettant de réaliser le diagnostic dans ce cas ont été décrites.
Mots clés: HYPOPHOSPHATASIE/génétique; HYPO-PHOSPHATASIE/métabolisme; RADIOGRAPHIE/méthodes; PHOSPHATASE ALCALINE.
Referencias bibliográficas
1. Smith DW. Recognizable patterns of human malformation. 3 ed. Philadelphia: WB Saunders, 1982:284-5.
2. Waymire KG, Mahueren JD, Jaje JM, Guillarte TR, Caburn SP, Mac Gregor GR. Mice lacking tissue non – specific alkaline phosphatasa die from seizures due to defective metabolism of vitamin B-6. Nat genet 1995;11(1):45-51.
3. Mc Kusick VA. Mendelian inheritance in man. Catalog. 6 ed. Baltimore: Johns Hopkins Press, 1983:298,780.
4. Tekinalp G, Gurakan B, Yalgin S, Caglar M, Ergin H. Neonatal form of hypophosphatasia. A case report. Turk J Pediatr 1995;37(4):421-4.
5. Murray RO, Jacobson HG. Radiología de los trastornos esqueléticos. 2 ed. La Habana: Editorial Científico-Técnica, 1982:1082-3.
6. Nelson WE, Behrman RE, Vaughan VC. Tratado de pediatría. 1 ed. La Habana: Editorial Científico-Técnica, 1988:1714-22.
7. Gabay L, Leiba J. Primary hypophosphatemic rickets: an uncommon cause of bowed legs. West Indian Med 1990;39(3):186-9.
8. Tekinalp G, Yukselen A, Balkansi F, Coskunt YM. Hypophosphatasia a newborn infant. Turk J Pediatr 1995;37(1):61-5.
9. Machtei EE, Ben-Yehouda A, Zubery Y, Sela RA. Lack of evidence for hypophosphatasia as a factor in the pathogenesis of early onset periodontitis. J West Soc Periodontal 1994;42(4):113-7.
10. Altay AN, Kocadereli Y, Atar G. Palatal expansion with a total denture. J Clin Pediatr Dent 1995;19(4):251-3.
11. Caffey J. Diagnóstico radiológico en pediatria. 7 ed. La Habana: Editorial Científico-Técnica; 1982:1437-8.
Recibido: 24 de diciembre de 2000. Aprobado: 24 de enero de 2001.
Dra. Elsa Luna Ceballo. Calle 12 No. 1707 e/n 17 y 19. Bolondrón. Matanzas. Cuba.
1 Especialista de I Grado en Genética Clínica.
2 Especialista de I Grado en Radiología.
3 Especialista de II Grado en Ortopedia y Traumatología. Profesora Consultante.

