










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Scheuthauer Marie Sainton fue descrito por primera vez en 1766 por Moran y, posteriormente, en 1897 por Pierre Marie y Sainton, quienes la nombraron disostosis cleidocraneal. Es también conocida en la literatura médica con el nombre de enfermedad de Marie Sainton, síndrome de Scheuthauer Marie Sainton y disostosis mutacional.
La disostosis cleidocraneal es una enfermedad poco frecuente, que sigue un patrón de herencia autosómico dominante con expresividad variable y es común en varios miembros de una misma familia. Se define por una serie de alteraciones típicas,1,2 entre las que se encuentran la ausencia total o parcial de clavículas, alteraciones óseas craneales, facie característica (hipertelorismo, seudoprognatismo), malformaciones dentales y hipoacusia de transmisión.
La mutación más frecuente se encuentra en el gen CBFA1/RUNX2, localizado en el cromosoma 6p216-11.3,4 Existe una correlación entre el fenotipo y el genotipo. Sin embargo, también se han descrito casos esporádicos.2,4 Afecta en la misma proporción a hombres y mujeres.3,5 Los pacientes afectados por este trastorno presentan agenesia o disgenesia clavicular. El diagnóstico se realiza a través de la radiografía de tórax, la cual confirma la ausencia o formación incompleta de las clavículas,1,6 Además, se describe al examen físico tórax acampanado, el cual se evidencia en el período de recién nacido. En el estudio del cráneo,6,7 en el período neonatal, puede encontrarse suturas y fontanelas amplias, occipucio prominente, afectación del clivus y silla turca; cuerpo del esfenoides ensanchado y aplanado, además de presencia de huesos wormianos. El cierre de suturas y fontanelas es tardío. En los adultos con este síndrome se describe hipertelorismo, aplanamiento de la base de la nariz, hipoplasia del maxilar superior, prognatismo y persistencia de la sutura mentoniana. La malformación de los huesos del cráneo puede provocar hipoacusia8 (una complicación frecuente y que debe buscarse de forma dirigida). Es normal que la primera dentición aparezca de forma tardía, al igual que la definitiva. En la literatura también se describen anomalías dentales tanto en número como en localización y esmalte.8,9 A la altura de la pelvis existe retraso o ausencia en la osificación de las ramas del pubis e hipoplasia de las alas ilíacas. Dichas alteraciones predisponen a que la mayoría de las mujeres afectadas requieran cesárea en sus partos y así evitar futuras complicaciones obstétricas.4,6 No existe tratamiento específico para los problemas óseos,6,7 pero estos pacientes deben contar con seguimiento y evaluación periódica por traumatólogo, ya que dentro de las complicaciones descritas destacan la dislocación de hombros y artrosis en la etapa adulta. Al mismo tiempo, el cuidado dental debe ser prolijo, se requiere de una evaluación dental desde el comienzo.10,11,12 Si existe el antecedente familiar o personal de disostosis cleidocraneal, es fundamental recibir consejo genético por el especialista.4,13
El diagnóstico diferencial se debe realizar con enfermedades como: pignodisostosis, crouzón o disostosis cráneo-facial y con síndromes como los de Richard Collins, Pierre Robin, Apert y Hollermann-Streiff y de Rubinstein-Taybi.14 El objetivo de esta presentación es describir las características específicas del Síndrome de Scheuthauer Marie Sainton en un paciente atendido en el Policlínico Docente “Efraín Mayor Amaro”.
Presentación de caso
Paciente masculino de 74 años de edad, nacido de parto distócico (cesárea), debido a que la madre presentó desproporción cefalopélvica. Acudió a la consulta de Medicina General Integral, del Policlínico Docente “Efraín Mayor Amaro”, municipio Cotorro, La Habana, con tos y secreción nasal. Fue el último hijo de cuatro embarazos, el primer hijo con antecedentes de salud aparentes, dos muertes fetales y el paciente en estudio que presenta varias alteraciones óseas y dentales, y se constató en él ausencia de ambas clavículas. Tenía como antecedentes patológicos personales la persistencia de dientes temporales y retraso en la erupción de los permanentes, retraso en el desarrollo pondoestatural. Dentro de los antecedentes patológicos familiares se evidenció el patrón hereditario de esta enfermedad, dado que madre e hija presentaron trastornos en la erupción dentaria y alteraciones esqueléticas (baja estatura y ausencia clavicular).
Al examen físico se observó paciente de raza blanca, de 1,56 cm de talla, peso 79 kg, índice de masa corporal: 32 obesidad grado I, miembro superior derecho 70 cm, miembro superior izquierdo 67 cm (antebrazos cortos y asimétricos), miembro inferior derecho 92,5 cm, miembro inferior izquierdo 90,5 cm, genus valgus bilateral, pie plano bilateral, tórax angosto (Fig. 1), fosas supra- e infraclavicular ausentes, hombros caídos, ausencia bilateral de clavículas que permite la unión de los hombros en línea media (Fig. 2). Además, se constató escaso esmalte dental propenso a caries con frecuencia.
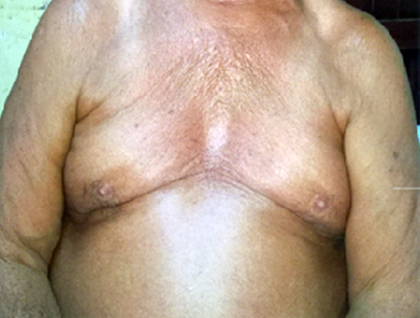
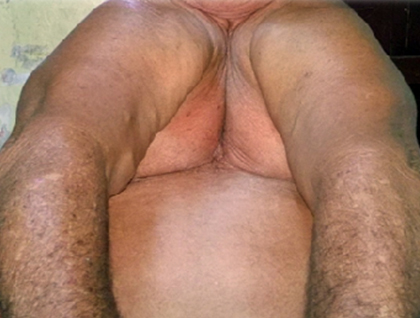
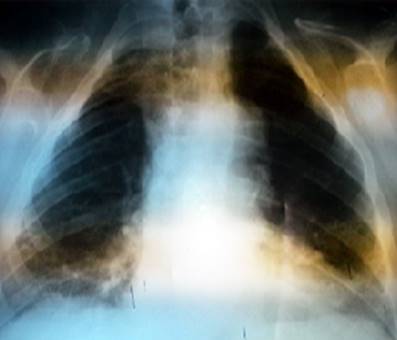
Se realizaron varios estudios para confirmar el diagnóstico como radiografía simple posteroanterior de tórax (Fig. 3), en la que se visualizó la ausencia de manera bilateral de las clavículas.
También, se realizó ultrasonido abdominal en busca de otras alteraciones. Se comprobó que ambos riñones tenían una buena relación cortico medular, parénquima sin alteraciones; no se define litiasis, ni dilatación. El riñón derecho medía 90 mm de largo, 45 mm de ancho y 9 mm de parénquima; y el izquierdo medía 98 mm de largo, 57 mm de ancho y 10 mm de parénquima. La próstata se encontraba heterogénea, redondeada de bordes regulares con calcificación central, con una medida de 49 mm de alto, 44 mm de ancho y 46 mm de largo, con un volumen de 32 cm3.
En cuanto al motivo de consulta se interpretó como un catarro común para lo cual se le indicó abundantes líquidos, analgésicos, antihistamínicos y Vitamina C. Para el tratamiento del síndrome de Scheuthauer Marie Sainton, el paciente derivado a la consulta de estomatología. Desde el punto de vista óseo, no se realizó ninguna cirugía correctora a voluntad del paciente.
Discusión
La displasia cleidocraneal es una enfermedad de herencia autosómica dominante, con penetrancia completa y expresividad variable, resultante de la mutación con pérdida de función del gen RUNX2 (runt-relatedtranscription factor 2; locus 6p21.1), un factor de transcripción esencial para la diferenciación de los osteoblastos y maduración de los condrocitos. La displasia cleidocraneal se caracteriza típicamente por la ausencia o hipoplasia de las clavículas, retraso en el cierre de las fontanelas y anomalías dentales, dichas características las presentaba el paciente de esta presentación.15 El gen RUNX2 es el único asociado a la displasia cleidocraneal. Entre 60 % y 70 % de los pacientes tienen una mutación identificable en el RUNX2. Cerca de 13 % de los casos presentan microdeleciones que incluyen RUNX2 y genes contiguos. También se han reportado rearreglos cromosómicos y deleciones que incluyen al locus 6p21.1, que se asocian con disrupción o déficit de RUNX2. Se han identificado más de 90 mutaciones en RUNX2. No hay diferencias fenotípicas significativas entre los pacientes con deleciones, mutaciones con corrimiento del marco de lectura o mutaciones de sentido adverso de RUNX2.16
El gen RUNX2 se expresa desde etapas tempranas del desarrollo embrionario17 y su principal función es encargarse de huesos y cartílagos con una fuerte expresión de carácter osteogénico y odontogénico. También se ha demostrado que regula la expresión de otros genes asociados con el desarrollo óseo, como el colágeno tipo 1, la osteocalcina, la osteopontina y la colagenasa.18 Algunos estudios sugieren que el gen RUNX2 regula la expresión de moléculas en el mesénquima y, a su vez, actúa recíprocamente en el epitelio dental para controlar su crecimiento y diferenciación. Esto explica, en parte, las anomalías dentales que se encuentran en pacientes con displasia cleidocraneal y en ratones heterocigotos para el gen RUNX2.16 La mutación tiene como consecuencia la haploinsuficiencia de RUNX2 y pobre diferenciación celular de los precursores osteoblásticos. La expresión de RUNX2 es constitutiva en la osteogénesis neoplásica, y la elevación en su expresión se ha asociado a la etiología del sarcoma. Su sobreexpresión en ratones predispone el desarrollo de linfoma de células T, osteopenia y fracturas múltiples; y en seres humanos, el desarrollo de craneosinostosis, hipoplasia maxilar y braquidactilia.19
Las principales manifestaciones clínicas de esta displasia son la aplasia o hipoplasia de las clavículas y la osificación insuficiente de la fontanela anterior. Las anomalías de las clavículas permiten el movimiento excesivo de los hombros, los que pueden ser aproximados anteriormente.11 En la mayoría de los casos, las anomalías claviculares son bilaterales; cuando es unilateral, las anomalías del lado derecho son las más comunes. En 10 % de los casos las clavículas están completamente ausentes. Las características faciales incluyen frente prominente, hipertelorismo, hipoplasia mediofacial, puente nasal deprimido y paladar alto.20 Las anomalías esqueléticas descritas son baja talla, falanges distales cortas, tórax estrecho en forma de cono, escoliosis, rodilla valga o pie plano; en las manos, las características son las epífisis cónicas.21 En el paciente de esta presentación lo que predominaba era el tórax angosto, genus valgus y el pie plano. Las anomalías dentales incluyen dientes supernumerarios en la dentición primaria y secundaria, que conduce a apiñamiento y mal oclusión, que se exacerba por la retención de dientes primarios. También se presenta retraso en la erupción de dientes permanentes e hipoplasia del esmalte,11) características similares a las del caso en cuestión.
Usualmente, la displasia cleidocraneal no se asocia a déficit cognitivo; sin embargo, en reportes recientes, Cheng y otros informaron de una paciente con deleción 6p21.2-p12.3, que incluía los genes CUL7, VEGFA, NFKBI y RUUNX2, quien además presentaba retraso en el desarrollo psicomotor y mala cicatrización de heridas.19Northup y otros reportaron una paciente con displasia cleidocraneal asociada a un rearreglo complejo del cromosoma 6 [der(6)ins(6)(p21.1q25.3q27)inv(6) (p25.3q27)], que resultó en disrupción del gen RUNX2, relacionado con alteraciones en el desarrollo psicomotor.15)
Radiológicamente, se puede encontrar un retraso en el cierre de las suturas sagital y metópica y de las fontanelas; así mismo, senos paranasales subdesarrollados o ausentes, defectos claviculares, escápulas pequeñas y deformadas, acortamiento de las falanges medias, alargamiento del segundo metacarpiano, defectos del arco neural cervicotorácico, acuñamiento posterior de las vértebras torácicas, espondilólisis lumbar, crestas iliacas hipoplásicas, retraso o ausencia de osificación del núcleo de la sínfisis púbica, ensanchamiento de la articulación sacroilíaca y cartílago trirradiado, cuello femoral varo, alargamiento de la cabeza femoral, calcáneos cortos, astrágalos estrechos y deformidades del antepié similares a las que aparecen en las manos.
No hay diferencias en la prevalencia de la enfermedad según etnia o género. La prevalencia se ha estimado en menos de un caso por millón, aunque esta puede estar subestimada, debido a que muchas personas con esta displasia no acuden a consultas médicas.20
El manejo debe ser individualizado, según las necesidades de cada paciente, derivadas de su fenotipo. Así, por ejemplo, el manejo quirúrgico de deformidades en clavículas con escisión del segmento clavicular será solo en caso de que estas causen problemas neurológicos, vasculares o en la piel. Las anomalías dentales son un aspecto importante en el tratamiento de estos pacientes: algunos requerirán múltiples tratamientos para extracción de dientes supernumerarios y el logro de un correcto alineamiento dental.
También es importante identificar la pérdida de audición, que puede deberse a cambios estructurales y funcionales del hueso temporal y huesos del oído, junto con la formación inusual o anormal del paladar o disfunción de la trompa de Eustaquio, que puede requerir la colocación de tubos de ventilación; sin embargo, también la hipoacusia puede deberse a la función anormal de los nervios auditivos. Por lo tanto, es preciso que un otorrinolaringólogo evalúe a los pacientes de manera rutinaria y que sean sometidos a pruebas audiológicas.
La cirugía craneofacial puede ser necesaria para corregir las anomalías del cráneo. El cuello femoral varo se trata con osteotomía femoral.22 En los casos en que se presenten con retardo mental o retardo del desarrollo psicomotor, es necesario descartar rearreglos cromosómicos a través del cariotipo convencional o con la utilización de la hibridación genómica comparativa, de acuerdo con el criterio clínico.23 La consejería genética de la displasia se basa en la herencia autosómica dominante, en la cual el riesgo para la paciente de tener un hijo afectado es de 50 %. Cabe aclarar que esta enfermedad varía en su presentación clínica y que sus principales complicaciones son de orden dental y otorrinolaringológicas, las cuales deben ser intervenidas de modo temprano. Además, pese a ser una enfermedad de carácter sistémico, tiene un buen pronóstico, y solo en casos esporádicos relacionados con rearreglos cromosómicos se asocia con déficit cognitivo; de ahí que la consejería genética deba ser parte del manejo integral de cada caso.12,24)
A modo de conclusión, las características específicas del síndrome de Scheuthauer Marie Sainton detectadas en el paciente fueron: el patrón heridatario de la enfermedad pues la madre y la hermana presentaban ausencia clavicular; además, presencia de tórax angosto, deformidades óseas como genus valgus y pie plano, persistencia de dientes temporales y retardo en la erupción de los permanentes. Se confirmó el diagnóstico con una radiografía de tórax.

