










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.13 n.1 Ciudad de la Habana ene.-jun. 2000
Hospital Pediátrico "Mártires de Las Tunas"
Síndrome Axenfeld-Rieger. Presentación de un caso
Dra. Daysi Flores Pérez,1 Dra. Yunia H. Labrada Rodríguez1 y Dra. Migdalis González Leyva1RESUMEN
Se presenta el estudio clínico de un caso representativo del Síndrome de Axenfeld-Rieger. En el examen clínico se constató corectopia, policoria, hipoplasia iridiana, glaucoma, hipertelorismo, aplanamiento de la base de la nariz, hipoplasia maxilar, alteraciones de la audición, dermatitis y retraso mental moderado. En esta paciente se realizó tratamiento quirúrgico, trabeculectomía en ambos ojos, por presentar cifras de presión intraocular elevadas.Descriptores DeCS: IRIS/anomalias; ENFERMEDADES DEL IRIS.
Este síndrome representa un espectro de desórdenes del desarrollo caracterizados por una línea de Schwalbe prominente desplazada en sentido anterior (embriotoxon posterior), con fibras de iris periférico unidas al embriotoxon posterior, hipoplasia del iris y disgenesia de la cámara anterior que conduce a glaucoma en el 50 % de los casos.1
Las condiciones referidas previamente acerca de la anomalía de Axenfeld, anomalía de Rieger y síndrome de Rieger frecuentemente se sobreponen, por lo que ahora son agrupadas en una simple entidad conocida como síndrome de Axenfeld-Rieger.1
El iris puede ser transiluminable. La hipoplasia de iris puede fluctuar desde un adelgazamiento ligero del estroma hasta la marcada atrofia con formación de agujeros, corectopia y ectropión uveal. La megalocórnea y microcórnea pueden ocurrir.
En ocasiones, se asocia a defectos de los dientes y huesos faciales, piel periumbilical redundante, hipospadias y anomalías de la glándula pituitaria.1
Relato del caso
Escolar de 13 años de edad, femenina, blanca, que acude a consulta por disminución de la visión y presencia de varias pupilas en ambos ojos. Presenta antecedente patológico personal de asma bronquial y trastornos del aprendizaje. Recogemos como dato de interés la existencia de consanguinidad (padres primo-hermanos). Al realizar el examen oftalmológico apreciamos hipertelorismo y aplanamiento de la base de la nariz. La agudeza visual es de 0,2 OD y 0,1 OI que no mejora con cristales. Presenta corectopia, policoria e iris hipoplásico en ambos ojos, con alteraciones más marcadas en el OI.
La biomicroscopia mostró en el OD; embriotoxon posterior, corectopia, policoria e hipoplasia iridiana; en el OI: embriotoxon posterior, corectopia, hipoplasia iridiana con zonas de atrofia marcada.
Fondoscópicamente encontramos papilas pálidas, con excavaciones 0,6 OD,. 0,8 OI y rechazamiento nasal de vasos. Se detectó en el campo visual aumento de la mancha ciega OD y reducción concéntrica del campo visual a 15º OI.
En la gonioscopia observamos bandas de tejido que se extienden desde el iris periférico hasta una prominente línea de Schwalbe y una inserción alta del iris, en la porción posterior de la malla trabecular.
Las presiones intraoculares en ambos ojos eran elevadas, 36 mm HgOD y 42 mmHg OI por lo que decidimos realizar trabeculectomía en ambos ojos. Actualmente la niña presenta presiones intraoculares normales y evoluciona favorablemente.
En el examen clínico general encontramos que presenta alteraciones de la audición con audiometrías alteradas, implantación baja de las orejas, hipoplasia maxilar, dermatitis de etiología no precisada y retraso mental.
Discusión del caso
En nuestro caso la paciente presenta manifestaciones oculares y generales del síndrome de Axenfeld-Rieger, (fig.) entidades que actualmente se unifican por tener patogenia idéntica: migración anómala de las células de la cresta neural que explica la persistencia de los vestigios de endotelio en la cara anterior del iris y la adherencia del iris en la línea de Schwalbe, así como anomalías extraoculares dentofaciales.2 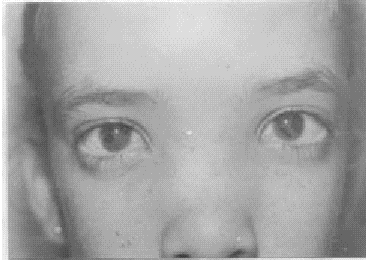
FIG. Síndrome Axenfeld-Rieger.
El iris es anormal en ojos con síndrome de Axenfeld-Rieger, pero existen líneas de Schwalbe desplazadas en sentido anterior con procesos iridianos periféricos, éste es el síndrome de Axenfeld. Si los defectos del iris están presentes, es clasificado como anomalía de Rieger. La patología del iris puede extenderse desde un suave adelgazamiento del estroma (iris hipoplásico) hasta la marcada atrofia con formación de orificios, corectopia y ectropion uveal. Cuando la corectopia está presente, la pupila es desplazada hacia la adhesión iridocorneal. La atrofia y formación de agujeros típicamente ocurren en el cuadrante alejado de la dirección de la corectopia.2,3
Otras anomalías oculares pueden ocurrir con frecuencia que incluyen: estrabismo, catarata, quistes dermoides, desprendimiento de retina, degeneración macular, colobomas coriorretinianos, hipoplasia coroidea e hipoplasia de la cabeza del nervio óptico.2
El glaucoma es posible que se manifieste durante la infancia, más comúnmente en la infancia tardía3 como es el caso de nuestra paciente; se produce a causa del desarrollo incompleto de la malla trabecular y canal de Schlems o por cierre angular secundario a sinequias. El glaucoma aparece más frecuente en pacientes con cambios iridianos centrales y en aquellos con pronunciada inserción anterior periférica del iris en la malla trabecular.
El glaucoma es difícil de controlar y causa pérdida significativa de la visión en un alto porcentaje de pacientes. La trabeculectomía es el procedimiento de elección en estos casos.2-5
Este síndrome se asocia con muchas anomalías sistémicas, en particular aquellas que envuelven defectos del desarrollo de los dientes y huesos faciales. Los defectos dentales pueden incluir reducción en el tamaño (microdontia), una disminución en el número y dientes espaciados (hipodontia) y ausencia focal de dientes (oligodontia o anodontia). Las anomalías faciales pueden incluir hipoplasia maxilar con aplanamiento de la cara media, inclinación del labio superior y prominente labio inferior. Hipertelorismo, telecantus y nariz ancha y aplastada. 2,5
Otras alteraciones menos frecuentes oueden asociarse como deficiencia de la hormona del crecimiento y corta estatura, trastornos del corazón, del oído medio, deficiencia mental, al binismo oculotáneo y variedad de desórdenes neurológicos y dermatológicos.
SUMMARY
A clinical case report is presented, which is representative of Axenfeld-Rieger syndrome. Clinical examination demonstrated corectopia, polycoria, radial hypoplasia, glaucoma, hypertelorism, flattening of nose bases, maxil larynhypoplasia, hearing disorders, dermatitis, and moderate mental retardation. This patient was undergoes to surgical treatment, trabeculotomy in both eyes, because of high figures of intraocular pressure.Subject headings: IRIS/ abnormalities; IRIS DISEASES.
REFERENCIAS BIBLIOGRÁFICAS
1. Slamonts TL. Pediatric ophthalmology and strabismus. Basic and clinical science course. Section 6. San Francisco: American Academy of Ophthalmology, 1994;95:59-66.
2. Wright KW, Buckley EG, Del Monte MA. Pediatric ophthalmology and strabismus. St. Louis: Mosby, 1995:303-6.
3. Kanski JJ. Oftalmología clínica 3 ed. Barcelona: Mosby / Doyma Libros, 1996:215.
4. Kanski JJ, Mc Allister. Glaucoma. Barcelona: Gráficas Catalanas, 1991:82-3.
5. Ritch R, Shields MB, Krupin T. The glaucomas. 2 ed. St. Louis: Mosby, 1996:875-88.
Recibido: 22 de noviembre de 1999. Aprobado: 16 de diciembre de 1999.
Dra. Daysi Flores Pérez. Calle Eddy Martínez, Edificio 30, apto. C-8, entre A. Cevereco y Línea, Buena Vista, Las Tunas.

