










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión impresa ISSN 0864-2176versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.16 n.1 Ciudad de la Habana ene.-jun. 2003
Hospital General Docente "Ernesto Guevara de la Serna"
Rasgos epidemiológicos de ciegos y débiles visuales por retinosis pigmentaria
en la provincia Las Tunas
Dra Luisa González Hess,1 Dra. Elizabeth Ramírez Pérez,2 Dra. Rosa M. Pérez Guerrero3 y Dra Adriana Abreu Leyva2
Resumen
Se estudiaron 325 pacientes con el diagnóstico retinosis pigmentaria en la provincia Las Tunas. De ellos, se encontró que el 24,9 y el 10 % eran ciegos y débiles visuales respectivamente, donde se obtuvo una tasa de ciegos por retinosis pigmentaria de 1,45 x 10 000 habitantes y de débiles visuales de 0,6 x 10 000 habitantes. El tipo de herencia que predominó en ambos grupos fue la autosómica recesiva.
DeCS: RETINITIS PIGMENTOSA/epidemiología; CEGUERA/etiología; CEGUERA/epidemiología.
La función de la retina es detectar la luz y enviar los estímulos apropiados para la respuesta cerebral. Las enfermedades hereditarias, que causan degeneración en la retina, como la retinosis pigmentaria (RP) y la degeneración macular, así como algunas formas raras de enfermedad retiniana, llevan a quienes la padecen a la ceguera total o parcial.1-3
La RP se define como un grupo de enfermedades hereditarias caracterizadas por la pérdida de los fotorreceptores y de la función del epitelio pigmentario de la retina.4 Su prevalencia en los EE.UU., en Francia y en Cuba es de 1 x 4 000, 1 x 4 900 y 1 x 4 082 habitantes, respectivamente. La provincia Las Tunas mostró una frecuencia de 1 x 2 428 habitantes.5
La pérdida visual por RP es importante hacia la tercera década de la vida, con implicación en el plano social y personal del paciente. La integridad del individuo se basa en la relación de equilibrio entre los aspectos psicológicos, intelectuales y corporales y con la enfermedad se rompe la estabilidad y el órgano visual se convierte en el centro de atención del paciente.6
La RP ocupa un lugar importante en todas partes del mundo. La existencia de un programa nacional en Cuba, permite conocer el estado de cada uno de los pacientes. Independientemente de las posibilidades de prevención implícitas. De esta manera se hizo factible la realización del estudio de ciegos y débiles visuales por esta causa en la provincia, lo que contribuirá al conocimiento de su influencia dentro de las causas de ceguera en el país.
Métodos
Se realizó un estudio epidemiológico descriptivo en el período comprendido de enero de 1996 hasta diciembre de 2001. Se eligió como muestra los 325 pacientes registrados y clasificados en la provincia Las Tunas, con el diagnóstico de RP en cualquiera de sus formas clínicas.
Para establecer el diagnóstico de ciego y débil visual, se tomaron en consideración los parámetros establecidos por la Organización Mundial de la Salud (OMS), que incluyen el concepto de ceguera como una alteración ocular, en la que el paciente posee una agudeza visual inferior a 0,1 en el menor ojo con corrección óptica o campo visual no superior a los 10 grados y débil visual, a aquellos con visión hasta 0,3 en el menor ojo con corrección óptica.6
Se realizó examen oftalmológico completo y se hizo énfasis en la determinación de la agudeza y campo visual.
Para la recogida de datos se utilizó la historia clínica epidemiológica diseñada por el grupo nacional de RP, donde se recoge la información general (sexo, edad y nombre) y específicos (debut, herencia, formas clínicas y enfermedades asociadas). Los específicos se basan en la clasificación cubana de RP del Dr. Orfilio Peláez Molina (Experiencia cubana en la retinosis pigmentaria. Primer Simposio Internacional de RP, La Habana, 1994). Se agregó la condición de ciego, débil visual y excluido de ambas categorías.
Los datos fueron computados por el sistema ACCES y se utilizó la prueba de hipótesis para canalizar los resultados, se consideraron significativas las cifras de p < 0,05 y altamente significativas a las de p < 0,001.
Resultados
De los 325 pacientes diagnosticados con RP, el 24,9 % son ciegos y el 10,5 % son débiles visuales. Estos resultados reportaron un predominio de los ciegos. Al determinarse el total, se encontraron 115 pacientes incapacitados visuales por esta enfermedad para un 35,4 %.
Con respecto al sexo, como se presenta en la figura 1, se encontró un número mayor en el masculino, con 53,1 % en los ciegos y un 61,8 en los débiles visuales y en el sexo femenino un 46,9 % de ciegos y un 38,2 de débiles visuales.
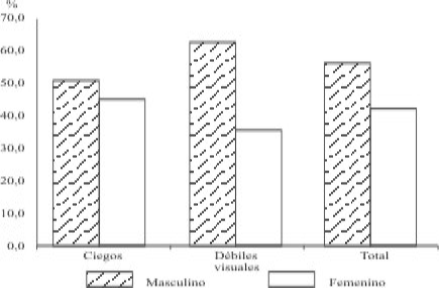
Fig. 1. Distribución por sexo de ciegos y débiles visuales.
El comportamiento de la enfermedad por grupos de edades se observa en la figura 2 mostró predominio de los pacientes ciegos en el grupo comprendido entre los 40 y los 60 años con un 33 %, mientras que en los débiles visuales fue de 16 a 30 años con el 35,3 %.
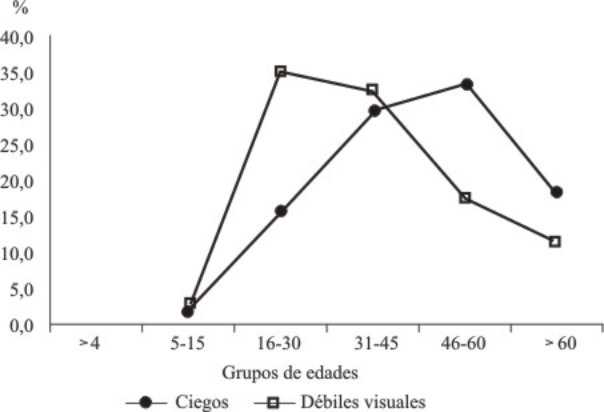
Fig. 2. Frecuencia según grupos de edades.
La figura 3 de este estudio muestra predomino de la herencia autosómico recesiva con un 38,3 % de ciegos y un 38,2 % de débiles visuales, seguida de la no definida con un 29,6 y un 32,3 % de ciegos y débiles visuales respectivamente. La ligada al X fue la que reportó la menor cantidad de casos con 4,9 % de ciegos y 8,8 % de débiles visuales, cifras que coinciden con los reportes anteriores.
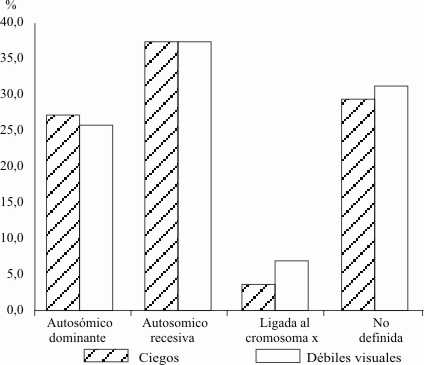
Fig. 3. Distribución según tipos de herencia.
Al relacionar el debut de la enfermedad con la presencia de ciegos y débiles visuales como se muestra en la figura 4, se encontró una mayor cantidad de pacientes con debut precoz para un 63,0 % de ciegos y un 52 % de débiles visuales, seguido del juvenil en menor cuantía el tardío.
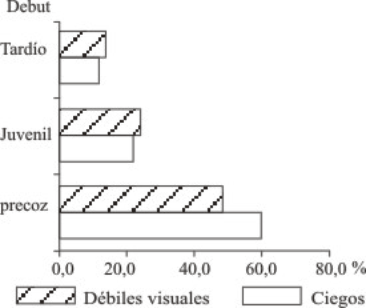
Fig. 4. Distribución según debut.
En relación con las formas clínicas se encontró que la forma típica predominó con un 85,4 % de ciegos y un 79,4 de débiles visuales, seguido de las asociadas con 8,6 % de ciegos y 14,7 de débiles visuales, donde fueron los síndromes de Usher y el Kearny Sayre los reportados. El Kearn Sayre es mucho menos frecuente, pero en este caso se detectó una familia con varios de sus miembros afectados.
Discusión
La RP es una distrofia retiniana que lleva de forma progresiva al deterioro visual. En este estudio las cifras de ciegos y débiles visuales por la enfermedad se consideran significativas, lo que se corresponde con otros reportes.6,7 De forma general la tasa de ciegos en la provincia fue de 1,45 x 10 000 habitantes, dato a tener en consideración si se conoce que la tasa de la enfermedad en Las Tunas es de 6,9 x 10 000 habitantes.
Según la OMS, la prevalencia de RP, en relación con el sexo, alcanza tasas estandarizadas con predominio en el masculino, de igual manera se comportaron en otros trabajos.4,8 En este estudio resultó un índice masculino femenino de 1,3 para los ciegos y de 1,6 para los débiles visuales.
Al analizar el comportamiento de la enfermedad por edades, es importante señalar que en el grupo de mayores de 60 años existe un menor porcentaje ya que existen pocos pacientes de esa edad con la enfermedad. Además, se observa una curva descendente en el caso de los débiles visuales a partir del grupo de 16-30 años, mientras en los ciegos, la curva es ascendente (fig.3). Esto se explica por el hecho de ser progresivo el deterioro visual. Peláez concluyó que existe una relación directa entre los años de evolución de la enfermedad y la agudeza y campo visual, donde la visión periférica se afecta de forma más severa y precoz.9 Otros señalan la presencia de RP aumenta con la edad. La poca cantidad de casos por debajo de los 15 años está relacionada con diversas causas, entre ellas el hecho de que el debut puede aparecer en etapas avanzadas, factor condicionado por el modo de herencia.10
Numerosas investigaciones muestran la gran heterogeneidad genética de esta enfermedad, que presenta todos los patrones de herencia mendeliana, casos esporádicos y últimamente, se habla también de la herencia mitocondrial.10,11 La herencia mitocondrial se descubre a la luz del desarrollo de la genética molecular, y en el caso de la RP, se relaciona con síndromes como el Kearn Sayre. Por otra parte se ha demostrado que en la herencia autosómico dominante se producen mutaciones de la rodopsina para lo cual no existe ningún tratamiento disponible.11 La herencia ligada al X es la de peor pronóstico y la menos frecuente. En cambio, la autosómico recesiva se describe como el patrón hereditario más frecuente de la RP.
Existen razones bien argumentadas que llevan a pensar que todo proceso crónico avanza más o menos lentamente en el transcurso de los años.9 O sea mientras más precoz aparece la retinosis, más posibilidades tiene de repercutir sobre las estructuras afectadas.
Existen diversas clasificaciones de la variedad clínica de la enfermedad Dr. Orfilio Peláez Molina (Experiencia cubana en la retinosis pigmentaria. Primer Simposio Internacional de RP, La Habana1994). En este caso se utilizó la clasificación cubana basada en las formas clínicas de la enfermedad y la define en forma típica que se caracteriza por hallazgos fondoscópicos como papila cérea, pigmentos en forma de osteoclastos y estrechez vascular, además de la alteración del ERG y CV, en forma atípica que incluye la sinc pigmento, la pravenosa, entre otras, y la asociada a síndrome como el Usher, el Kearn Saire entre otro, pero es el síndrome de Usher, el que con mayor frecuencia se asocia a la RP.12
Con este estudio se demuestra que la RP no sólo es una enfermedad de la retina que atañe exclusivamente al oftalmólogo, sino a la sociedad en general, por el efecto lesivo que sobre la visión es capaz de producir.
Conclusiones
La tasa de ciegos y débiles visuales por RP en la provincia mostró cifras significativas. Se demostró el predominio de la enfermedad tanto en ciegos como en débiles visuales, en los grupos de edades superiores a los 15 años y la herencia autosómico recesiva presentó un mayor porcentaje para los ciegos y débiles visuales.
Summary
325 patients with the diagnosis of retinitis pigmentosa were studied in the province of Las Tunas. Of them, it was found that 24.9 % and 10 % were blind and visually handicapped, respectively. A rate of blind patients caused by retinitis pigmentosa of 1.45 x 10 000 inhabitants and of visually handicapped of 0.6 x 10 000 inhabitants was obtained. The type of inheritance predominating in both groups was the autosomal recessive inheritance.
Subject headings: RETINITIS PGMENTOSA/epidemiology; BLINDNESS/etiology; BLINDNESS/epidemiology.
Referencias bibliográficas
1. Sullivan LS,Daeger SP. Inherited retinal degeneration: genet and clinical heterogeneity. Mol Med Today 1996 Sep;2(9):380-6.
2. Bellous AR, Kass MA, Lichtenstein SB. Retina and vitreous basic and clinical science course. Section 12. San Francisco: American Academy of Ophthalmology; 2000-01:56-7.
3. Kansai J&J, Oftalmología clínica. 4 ed. Barcelona: Mosby Doyma Libros SA;1999:330-2.
4. Peláez MO. Retinosis pigmentaria. Experiencia cubana. 1 ed. La Habana:edit. Científico-Técnica;1997:36-47.
5. Palmero A. Principales causas de ceguera en la provincia de Sancti Spiritus. Rev Cub Oftalmol 1988;2:83-9.
6. Rosemberg T, Klee F. Current trends in newly registered Blindness in Denmark. 1996 Aug;74(4):395-8.
7. Honda Y. Retinitis pigmentosa is selected for one registered serious disease by Japanese Ministry Health and Welfare. Nipon Yanka Gakkai Zsshi 1996 Apr;100(4):255-66.
8. Haim M, Rosemberg Z. Retinitis pigmentosa and allied disorders in Denmark. Acta Ophthalmol Copenh 1993 Oct;71(5):597-65.
9. Peláez MO, Palmero GA, Pérez G. El deterioro visual y su relación con los años de evolución de la retinosis pigmentaria. Rev Cub Oftalmol 1991;4(1):66-71.
10. Vaughan D. Oftalmología general/ D. Vaughan, T. Asbury. 11 ed.México: El Manual Moderno;1997:226-7.
11. Kaplan J. Clinical and heterogeneity in retinitis pigmentosa, Springer Verlag. Hum Genet 1990;85:635-42.
12. Pieke DS, Van Aarem A. Genetic heterogeneity of Usher syndrome Type II in a deutsh population. J Med Genet 1996 Sep;33(9):753-7.
Recibido: 25 de septiembre de 2002. Aprobado: 11 de julio de 2003.
Dra. Luisa González Hess.Calle 10 No. 30, Pueblo Viejo, Jesús Menéndez, Las Tunas, Cuba. E-mail: Luisagh@cubasi.cu
1 Especialista de I Grado en Oftalmología. Hospital General Docente "Ernesto Guevara de la Serna".
2 Especialista de I Grado en Oftalmología. Centro Provincial de Retinosis Pigmentaria.
3 Especialista de II Grado en Oftalmología. Profesora Auxiliar. Centro Provincial de Retinosis Pigmentaria.

