










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.17 n.2 Ciudad de la Habana jul.-dic. 2004
Centro Provincial de Genética de Villa Clara
Heterogeneidad clínica y genética de la retinosis pigmentaria. Importancia del estudio familiar y del tratamiento precoz. A propósito de una familia
Dr. Rubén Rangel Fleites,1 Dr. Noel Taboada Lugo2 y Téc. Gladys Membrides Pérez3
Resumen
Conocida es la heterogeneidad genética de la retinosis pigmentaria considerada por diferentes autores como un grupo de enfermedades hereditarias que se deben a distintas mutaciones y se transmiten por diferentes patrones mendelianos de herencia que se manifiesta en general, con un cuadro clínico similar en sus características fundamentales. Notable es también su heterogeneidad desde el punto de vista clínico pues distintos genotipos se corresponden con distintos fenotipos, pero incluso entre los individuos afectos de la misma mutación pueden ser manifiestas las diferencias existentes en diversos aspectos clínicos de la enfermedad, por ello se reconoce por los investigadores dedicados a su estudio lo indispensable de un adecuado estudio familiar para un diagnóstico correcto, sobre todo desde el punto de vista genético. Se presenta una familia clasificada inicialmente como afecta de retinosis pigmentaria típica autosómica recesiva que luego de la pesquisa clínico familiar en cascada se reclasificó como de herencia autosómica dominante. Se evidenció la importancia del exhaustivo estudio familiar y del tratamiento quirúrgico precoz. Palabras clave: Heterogeneidad genética/retinosis pigmentaria; estudio familiar/diagnóstico; tratamiento; cirugía.
Se denomina retinosis pigmentaria (RP) a un conjunto de degeneraciones progresivas de carácter hereditario que, de manera difusa, afectan primariamente a la función de las células fotorreceptoras y al epitelio pigmentario de la retina.1-5
El 25 y el 30 % de las mutaciones que presentan los casos de RP autosómica dominante lo son a nivel del gen de la rodopsina que se localiza en el brazo largo del cromosoma 3; también han sido detectadas mutaciones en el brazo corto del cromosoma 6, en el gen de la periferina y en la región pericentral del cromosoma 8. En la actualidad se discute la existencia de un cuarto locus implicado en la RP.1
Las siguientes mutaciones en el brazo largo del cromosoma 3 han sido encontradas con frecuencia: glutamina-344-del, arginina-135-leucina, arginina-135-triptófano, treonina-17-metionina, treonina-58-arginina, prolina-23-histidina, prolina-347-leucina, leucina-46-arginina, leucina-68-del, arginina-69-del, treonina-70-del, prolina-71-del. En el brazo corto del cromosoma 6 se ha detectado la mutación serina-212-glicina.1,4,6
Distintos genotipos se corresponden con distintos fenotipos. Por ejemplo, la forma de RP con rodopsina prolina-347-leucina, presenta campos visuales (CV) y amplitudes en el electrorretinograma (ERG) menores que otros pacientes con RP autosómico dominante sin esta mutación.1,7
Existe heterogeneidad clínica incluso entre pacientes con una misma mutación, lo que sugiere la implicación de algún otro factor además del defecto genético en sí mismo.1
El 30 de noviembre de 1994, en el marco del Primer Simposio Internacional sobre Retinosis Pigmentaria, fue presentada en Ciudad de La Habana, la técnica quirúrgica revitalizadora del Prof. Orfilio Peláez. Esta técnica consiste en un transplante del tejido adiposo vascular orbitario, de forma pediculada, que se sitúa en el espacio supracoroideo y que por un mecanismo de angiogénesis contribuye a mejorar la función de los fotorreceptores aún activos.8
Presentación de la familia
Procedentes del Centro Nacional de Retinosis Pigmentaria (CPRP) acuden al centro el caso índice y una hermana (II-5) con diagnóstico de RP típica autosómica recesiva, para continuar su tratamiento, (ver genealogía en la fig. 1) ambos habían sido diagnosticados desde hacía varios años y operados de cirugía revitalizadora (CRV) temporal inicialmente y después en los cuadrantes nasales superiores y mantuvieron desde entonces, sin variación su agudeza visual (AV) y el CV.
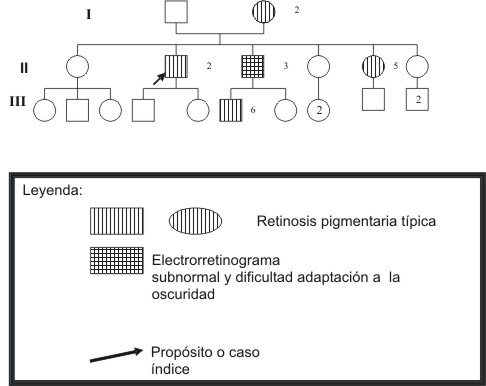
Fig. 1. Árbol genealógico de la familia.
A través del exhaustivo estudio familiar que incluyó la confección del árbol genealógico y la pesquisa clínica en cascada de la enfermedad realizado en el centro fueron diagnosticadas la madre del propósito (I-2) y un sobrino de éste con la enfermedad (III-6). De igual manera se examinó un hermano del caso índice, padre de este último caso, (II-3) que aún cuando no presentaba el cortejo de síntomas clínicos típicos, al examen físico se encontraron algunos hallazgos compatibles con la RP, lo que determinó que la familia fuera rediagnosticada como patrón de herencia autosómico dominante donde se evidencia la heterogeneidad clínica de la RP referido a la expresión fenotípica y a la edad de debut.
Principales características clínicas de los referidos individuos enfermos:
Caso II-2
Paciente MHV .
Debut (nictalopía): 11 años.
Diagnóstico positivo a los 25 años.
Inicia tratamiento CNRP: 48 años
Operado CRV temporal y nasal a los 50 y 52 años respectivamente.
ERG: extinguido.
Po: 16 mm Hg Ao.
AV: cdedo 2 ½ m Ao (indirecta).
CV: Fig. 2.
Se mantiene desde el tratamiento quirúrgico.
Resto del examen oftalmológico característico de RP típica. Estadio IV.

Fig. 2. Pericampimetría del caso II-2.
Caso II-5
Paciente JIHV.
Debut (nictalopía): 40 años.
Diagnóstico positivo a los 42 años.
Inicia tratamiento CNRP: 42 años.
Operado CRV temporal y nasal a los 44 y 46 años respectivamente.
ERG: extinguido.
AV: OI: cd 2 m y OD: 0.1 de diferencia
Po: 19 mm Hg Ao.
CV: Fig. 3.
Resto del examen oftalmológico característico de RP típica. Estadio IV.

Fig. 3. Pericampimetría del caso II-5.
Caso I-2
Paciente IVP.
Debut (nictalopía): 71 años.
Diagnóstico positivo a los 76 años.
No aceptó tratamiento quirúrgico. Inicia tratamiento a los 76 años.
ERG: inicialmente muy subnormal, actualmente extinguido.
Po 18 mm Hg Ao.
AV Inicial: OI: cd 30 cms y OD: cd 1 m.
Post CRV: OI: 0.2 y OD: 0.5
CV: Fig. 4.
Resto del examen oftalmológico característico de RP típica. Estadio III.
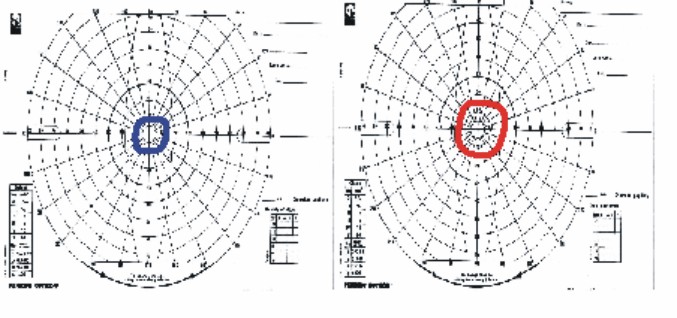
Fig. 4. Pericampimetría del caso I-2.
Caso III-6
Paciente MSCH.
APP ocular: miopía.
Debut (nictalopía): 15 años
Diagnóstico positivo a los 25 años.
Inicia tratamiento CPRP: 25 años.
Operado CRV temporal a los 28 años.
ERG inicial: subnormal Ao.
ERG 3 años después: extinguido.
Pericampimetría inicial: Fig. 5.
CV dos años después: Fig. 6.
AV inicial: OI: 0.5 y OD: 0.7
Post CRV: OI: 0.7 c/s/c y OD: 0.9 c/s/c.
Po: 19 mm Hg.
Resto del examen oftalmológico característico de RP típica. Estadio I.

Fig. 5. Pericampimetría del caso II-3.
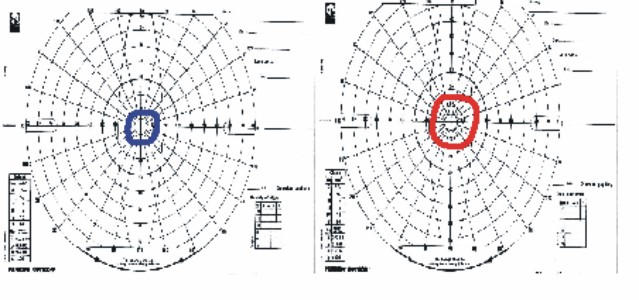
Fig. 6. Pericampimetría del caso III-6.
Caso II-3
Niega nictalopía. Dificultad a la adaptación a la oscuridad.
ERG: subnormal.
Pericampimetría: Fig. 7.
AV: AO: 1.0 c/s/c .
Examen oftalmológico sin alteraciones.

Comentario
La principal herramienta del genetista clínico lo constituye el examen físico y la exhaustiva caracterización familiar, ello puede resultar para algunos menos fascinante que la genética molecular, pero su accesibilidad y el impacto económico que supone su utilización, justifican que se vuelva la mirada hacia desde donde debe comenzar todo acto médico: la valoración cuidadosa de la clínica.9 En este caso se evidencia una vez más como el estudio clínico genético familiar resultó de vital importancia para un adecuado diagnóstico de la RP desde el punto de vista genético, toda vez que permitió reclasificar la familia desde el punto de vista genético, lo que reviste suma importancia, pues el primer elemento del asesoramiento genético (AG) es precisamente el diagnóstico, ya que en dependencia del patrón de herencia se establecen los riesgos de recurrencia para cada miembro familiar y permite brindar entonces un AG más objetivo.
Se constató la eficacia de la técnica quirúrgica cubana, al lograrse la detención del progreso del deterioro visual en los pacientes operados de CRV en estadios avanzados y la mejoría de la visión cuando éste fue aplicado en estadios mucho más precoces de la enfermedad, lo que justifica el diagnóstico precoz y el examen clínico en cascada en las familias, para la pesquisa de los individuos en riesgo, en dependencia del patrón de herencia con que se esté segregando la enfermedad, aún en los casos clínicamente asintómaticos.
En esta familia con RP autosómica dominante se observó una significativa diferencia en el inicio de la enfermedad en relación con el sexo, siendo su debut juvenil en los varones y muy tardío en las hembras lo que pudiera estar en relación con una mutación específica que determine esta forma de manifestación, no encontrada en otras familias con igual patrón de herencia.
Summary
It is known the genetical heterogeneity of retinitis pigmentosa considered by different authors as a group of hereditary diseases resulting from different mutations that are transmitted by different Mendelian patterns of inheritance that are generally manifested by a clinical picture that is similar in its fundamental characteristics. Its heterogeneity is also remarkable from the clinical point of view, since different genotypes correspond to various phenotypes, but even among the individuals affected with the same mutation the differences existing in dissimilar clinical aspects of the disease may be manifested. That's why, the investigators devoted to its study recognize that it is indispensable to conduct a family study to have a correct diagnosis, mainly from the genetical point of view. A family initially classified as affected by recessive autosomal typical retinitis pigmentosa that after the cascade clinical family screening was reclassified as dominant autosomal inheritance, was presented. The importance of the in-depth family study and of the early surgical treatment was evidenced.
Key words: Genetical heterogeneity/retinosis pigmentosa, family study/diagnosis; treatment; surgery.
Referencias bibliográficas
1. Gutiérrez Torre SM. Retinosis Pigmentaria. Clasificación y Tratamiento. 2001 URL disponible en: http://www.retinosis.org/old/librorp/inicio.htm
2. Peláez O, Herrera M, Mendoza MA, Paz G. Una década prodigiosa. Avan Médic de Cuba. 2001;25:28-31
3. Retinosis Pigmentaria. 2004 URL disponible en: http://www.retinosis.org/index.htm
4. Retinitis Pigmentosa. 2004. URL disponible en: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=omim
5. Grance L, Ponjavic V, Andreassson S. Full-field ERG, multifocal ERG and multifocal VEP in patients with retinitis pigmentosa and residual central visual fields. Acta Ophthalmol Scand. 2004 Dec;82(6):701-6.
6. Ayuso C, Baiget M, Palau F, Volpini V. Patología molecular hereditaria. En: Patología Médica II. Barcelona: ESPAXS, SA.2000. p. 1117-93
7. Wang DY, Chan WM, Tam PO, Lam DS, Chong KK, Fan BJ. Gene mutations in retinitis pigmentosa and their clinical implications. Clin Chim Acta. 2005 Jan;351(1-2):5-16.
8. Retinitis Pigmentosa. Técnica Quirúrgica Cubana. 1997 URL disponible en: www.sld.cu/instituciones/rp/retino.htm#tit4
9. Taboada N, Lardoeyt R. Criterios para el diagnóstico clínico de algunos síndromes genéticos. Rev Cubana Pediatr. 2003;75 (1). URL disponible en: http://bvs.sld.cu/revistas/ped/indice.html
Dr. Rubén Rangel Fleites. Centro Provincial de Genética de Villa Clara. Villa Clara, Cuba.
1Especialista de I Grado en Oftalmología. Centro Provincial de Retinosis Pigmentaria de Villa Clara.
2Especialista de I Grado en MGI y Especialista de I Grado en Genética Clínica. Centro Provincial de Genética de Villa Clara. Profesor Instructor ISCM de Villa Clara.
3Técnica en Optometría y Óptica. Centro Provincial de Retinosis Pigmentaria de Villa Clara.

