










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.17 n.2 Ciudad de la Habana jul.-dic. 2004
Centro Provincial de Genética Villa Clara
Atrofia óptica hereditaria autosómica dominante. A propósito de una familia
Dr. Noel Taboada Lugo,1 Dr. Roberto Lardoeyt Ferrer2 y Dr. Rubén Rangel Fleites3
Resumen
Entre las causas de pérdida insidiosa, bilateral y simétrica de la visión central se deben tener siempre presente las atrofias ópticas heredo degenerativas. La atrofia óptica hereditaria autosómica dominante es la forma más frecuente de atrofia óptica heredofamiliar simple o monosintomática. Se realizó la caracterización clínica de una familia con el diagnóstico de esta discapacidad visual.
Palabras clave: Atrofia óptica/herencia; Atrofia óptica/ simple o monosintomática.
Entre las causas de pérdida insidiosa, bilateral y simétrica de la visión central se deben tener siempre presente las atrofias ópticas (AO) heredo degenerativas. Establecer un diagnóstico etiológico diferencial de la AO en la infancia incluye las alteraciones hereditarias, metabólicas, comprensivas y estructurales. Las AO hereditarias constituyen un grupo especial de AO.1
Son de particular importancia, no solo por sus implicaciones genéticas, sino también por la frecuencia, con que ocurren. Pueden presentarse de forma aislada monosintomática, o bien asociadas a otras lesiones del SNC.1-3
La AO hereditaria autosómica dominante (OMIM no. 65500) 4 es la forma más frecuente de AO heredofamiliar simple o monosintomática, es producida por mutaciones en el gen OPA-1, con locus en 3 q28- q29, que se expresa específicamente en las células ganglionares y en las neuronas intrínsecas de la retina. Se han descrito más de 14 mutaciones en este locus.4-7
La disfunción visual es mucho más discreta en este trastorno que en la neuropatía óptica de Leber o en la AO recesiva.2
Presentación de la familia
En la fig. 1 se muestra el árbol genealógico de la familia.
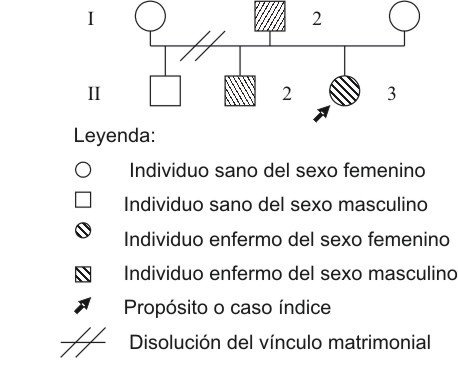
Fig. 1. Árbol genealógico de la familia.
A continuación se detalla la caracterización clínica de todos los individuos afectados en la familia:
Individuo I - 2:
Edad: 42 años.
Debut: infancia temprana.
AV. OD: 0.4/ OI : 0.6
Palidez papilar completa.
Pericampimetría: se muestra en la fig. 2.
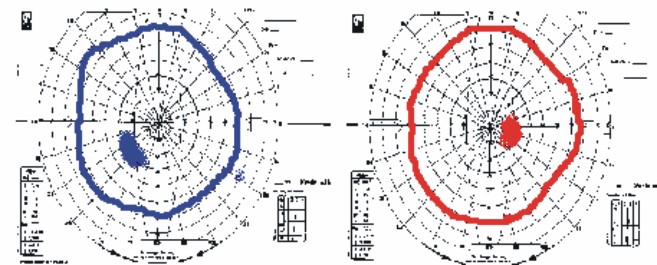
Fig. 2. Pericampimetría del individuo I - 2.
Individuo II -2:
Edad: 18 años.
Debut: 6 años.
AV. AO: 0.7
Palidez papilar temporal.
Pericampimetría: se muestra en la fig. 3.
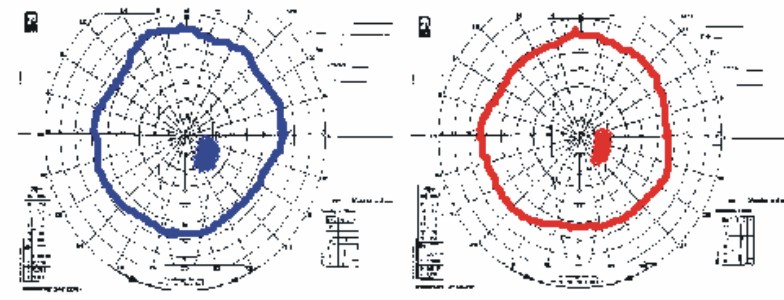
Fig. 3. Pericampimetría del individuo II- 2.
Propósito ( caso II -3)
Edad: 10 años.
Debut: 4 años.
AV. AO: 0.6
Palidez papilar temporal.
Pericampimetría: se muestra en la fig. 4.
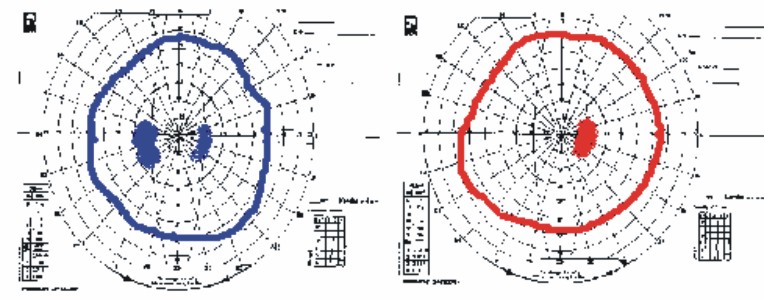
Fig. 4. Pericampimetría del individuo II- 2.
Comentario
Se pudo apreciar al examen físico que la papila es anormal en todos en todos los casos, con una palidez sectorial temporal en dos de ellos y una palidez completa en el otro. Este último era el paciente con mayor edad.
En el examen físico general realizado no se evidenció nistagmo, ni otra alteración neurosensorial en ninguno de ellos.
En las pruebas campimétricas, tal como se puede apreciar en las figuras, se encontró en todos los casos un discreto aumento de la mancha ciega, sin reducción de las isópteras del campo y escotoma cecal en un caso.
En el estudio de la visión cromática se evidenció en todos una discromatopsia del eje azul-amarillo.
Ante todo caso de AO bilateral en la infancia debe descartarse esta entidad, pues es la AO hereditaria más frecuente que existe,2,4 que por el tipo de transmisión mendeliana con que se segrega se encuentra entre los trastornos monogénicos con un riesgo de recurrencia alto (50 %) para la descendencia de los individuos afectados, por lo que el diagnóstico de esta entidad propiciaría un adecuado asesoramiento genético en las parejas o individuos de las familias donde se esté segregando el defecto, sobre todo por tratarse de un trastorno donde no existe la disponibilidad de un diagnóstico prenatal.
Summary
Among the causes of insidious, bilateral and symmetric loss of the central vision, the hereditary and degenerative optical atrophies should always be taken into account. The dominant autosomal hereditary optical atrophy is the most frequent form of simple or monosymptomatic hereditary family optical atrophy. The clinical characterization of a family with the diagnosis of this visual impairment was made.
Key words: Optical atrophy/inheritance; optical atrophy/simple or monosymptomatic.
Referencias bibliográficas
1. Atrofia óptica. 2005. URL disponible en: www.orpha.net/static/ES/atrofia_optica.html
2. Atrofia Óptica Autosómica Dominante. 2001. URL disponible en: www.alconlabs.com/ar/aj/new/2001/N0124.jhtml
3. Buono LM, Foroozan R, Sergott RC. Unexplained visual loss. Surv Ophthalmol. 2003; 48 (6):626-30.
4. Atrofia óptica autosómica dominante. URL disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=165500
5. Pesch UE, Fries JE, Bette S, Kalbacher H, Wissinger B, Alexander C, et al. OPA1, the disease gene for autosomal dominant optic atrophy, is specifically expressed in ganglion cells and intrinsic neurons of the retina. Invest Ophthalmol Vis Sci. 2004; 45 (11):4217-25.
6. Baris O, Delettre C, Amati-Bonneau P, Surget MO, Charlin JF, Catier A, et al. Fourteen novel OPA1 mutations in autosomal dominant optic atrophy including two de novo mutations in sporadic optic atrophy. Hum Mutat. 2003; 21 (6):656.
7. Payne M, Yang Z, Katz BJ, Warner JE, Weight CJ, Zhao Y, et al. Dominant optic atrophy, sensorineural hearing loss, ptosis, and ophthalmoplegia: a syndrome caused by a missense mutation in OPA1. Am J Ophthalmol. 2004; 138 (5):749-55.
Recibido: 14 de septiembre de 2004. Aprobado: 16 de noviembre de 2004.
Dr. Noel Taboada Lugo. Centro Provincial de Genética Villa Clara. Villa Clara, Cuba.
1Especialista de I Grado en Medicina General Integral y Especialista de I Grado en Genética Clínica. Instructor ISCM Villa Clara.
2Especialista de I Grado en Genética Clínica. Centro Nacional de Genética Médica. Ciudad de la Habana.
3Especialista de I Grado en Oftalmología. Centro Provincial de Retinosis Pigmentaria "Dr. Orfilio Peláez" Villa Clara.

