










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.22 n.1 Ciudad de la Habana ene.-jun. 2009
PRESENTACIÓN DE CASO
Síndrome de Noonan. Presentación de dos casos
Noonan's syndrome. Presentation of two cases
Lucy Pons CastroI; Teresita de J. Méndez SánchezII; Rosa Maria NaranjoII; Alejandro Arias DíazI; Mavys Soto GarcíaI; Mirta Silveira SimónI
I Especialista de I Grado en Oftalmología. Instituto Cubano de Oftalmología "Ramón Pando Ferrer", La Habana, Cuba.
II Especialista de II Grado en Oftalmología. Instituto Cubano de Oftalmología "Ramón Pando Ferrer", La Habana, Cuba.
RESUMEN
Se presentan las características oftalmológicas y clínicas de dos pacientes hermanos (hembra y varón) con diagnóstico del síndrome de Noonan. Este es un trastorno genético que produce desarrollo anormal de múltiples partes del cuerpo. Se caracteriza por una serie de signos y particularidades físicas que pueden variar ampliamente en rango y severidad según los casos. Generalmente se transmite como un rasgo genético autosómico dominante. Los casos que presentamos se caracterizan por: estenosis valvular pulmonar, hipertelorismo, retardo mental moderado, aspecto típico de la cara con filtrum (surco vertical en el centro del labio superior), párpados gruesos, epicanto, exoftalmos y ptosis palpebral.
Palabras clave: Síndrome de Noonan/etiología, genética, anomalías múltiples, manifestaciones oculares.
ABSTRACT
The ophthalmological and clinical characteristics of two sibling patients (male and female) diagnosed with Noonan´s syndrome were presented in this paper. This is a genetic disorder that causes abnormal development of many parts of the body. It is characterized by a series of signs and physical peculiarities that may widely vary in range and severity from one case to another. Generally, it is transmitted as a dominant autosomal genetic trait. The two cases had the following features: pulmonary valve stenosis, hypertelorism, moderate mental retardation, typical aspect of the individual's face with filtrum (vertical sulcus located in the center of the upper lip), thick eyelids, epicanthus, exophthalmos and palpebral ptosis.
Key words: Noonan's syndrome/etiology, genetics, multiple anomalies, ocular manifestations.
El síndrome de Noonan fue descrito en 1963 por Noonan y Ehmke en pacientes con estenosis valvular pulmonar, asociado a baja estatura, hipertelorismo y retardo mental moderado, entre otras alteraciones. La virtud de Jacqueline Noonan fue que además de indicar los signos clínicos mayores, observó que la cardiopatía más frecuente era la estenosis pulmonar (17 en 19 pacientes, Noonan, 1968), lo que diferenciaba a esta enfermedad del síndrome de Turner, donde la cardiopatía que se presenta con mayor frecuencia es la coartación de la aorta.1,2,3
El síndrome de Noonan afecta al menos a 1 de cada 2 500 niños. A diferencia del síndrome de Turner, los afectados por el Noonan no tienen alteraciones del cariotipo, es decir, no tienen dificultades con el número ni la organización de los cromosomas, estructuras que contienen la información genética. Diferentes estudios detectaron familias en donde el síndrome aparecía en varios miembros, bajo una transmisión vertical, con rasgos diferentes de unos a otros e incluso con generaciones saltadas (dominancia irregular), pero con un predominio de la herencia por vía materna. Esto estableció un patrón de herencia autosómica dominante, lo que significa que en estos casos sería necesaria la presencia de una mutación, ya en los genes de uno de los progenitores, y que esta mutación sería transmitida a los hijos con un 50% de probabilidad. Aunque en esta enfermedad la probabilidad de que una vez presente, se exprese de una forma grave sería del 14%. El hecho de que algunos niños no tengan un padre con el síndrome de Noonan refleja la posibilidad de una aparición esporádica, es decir, la presencia presumible de una nueva mutación, no presente en los genes de los padres.1,4
Desde hace unos pocos años se ha identificado el locus donde se ubica el gen que condiciona el fenotipo de al menos un gran porcentaje de personas con el síndrome de Noonan y que se sitúa en 12q 24 (Jamieson). En este emplazamiento del mapa genético se encuentra el primer gen específico identificado como posible responsable del síndrome de Noonan, denominado PTPN11.1 También puede ser causado por anomalías en los genes KRAS y PTPN11. Aproximadamente la mitad de las personas afectadas por este síndrome tienen una mutación en este último gen. Los individuos con una anomalía en el gen KRAS presentan una forma atípica o severa del síndrome de Noonan. Los problemas con estos genes hacen que ciertas proteínas involucradas en el crecimiento y desarrollo se vuelvan hiperactivas.5
Este único gen no puede explicar todos los casos, por lo que se espera el descubrimiento de otros genes que producen este síndrome.4,6
Un examen clínico cuidadoso, el análisis de cromosomas y el reconocimiento físico pueden ayudar al diagnóstico correcto, aunque este diagnóstico es fundamentalmente clínico: se diagnostican como tales a todos aquellos que cumplan las características fenotípicas que definen a este síndrome,1 el cual puede afectar tanto a varones como a hembras.7
PRESENTACIÓN DE CASOS
I. Paciente femenina de 6 años de edad, con diagnóstico por genética de síndrome de Noonan, a la edad de 2 años.
Antecedentes patológicos familiares: Madre con síndrome de Noonan.
Antecedentes patológicos personales: Agenesia vesicular y estenosis pulmonar. Esta última es un signo sumamente importante por la alta frecuencia de presentación y que permite entonces, diferenciar esta entidad del síndrome de Turner cuando existen dudas entre ambos.
Acude a consulta de Oftalmología Pediátrica por presentar ptosis palpebral bilateral más acentuada en el ojo derecho, con esodesviación e hipertrofia alternante (fig. 1). (Se obtuvo el consentimiento de los familiares para publicar estas fotos).

Al examen oftalmológico se encontró además, miopía de - 6,00 dioptrías en el ojo derecho con ambliopía profunda de este mismo ojo, epicanto, inclinación palpebral antimongoloide, proptosis, puente nasal poco desarrollado e hipertelorismo.
Encontramos otras alteraciones como la implantación baja de las orejas y el hirsutismo (fig. 2).

II. Paciente masculino de 3 años de edad, con diagnóstico por genética de síndrome de Noonan, a la edad de 1 año.
Antecedentes patológicos familiares: Madre con síndrome de Noonan.
Antecedentes patológicos personales: Estenosis pulmonar valvular con gradiente de 21 milímetros.
Acude a consulta de Oftalmología Pediátrica por presentar ptosis palpebral y exoftalmo bilateral más acentuado en el ojo izquierdo (fig. 3).
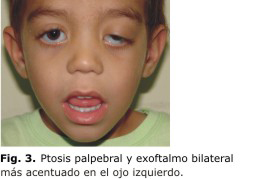
Al examen oftalmológico encontramos además, miopía de -10,00 dioptrías en el ojo izquierdo con ambliopía profunda de este mismo ojo, epicanto, inclinación palpebral antimongoloide, proptosis y puente nasal poco desarrollado.
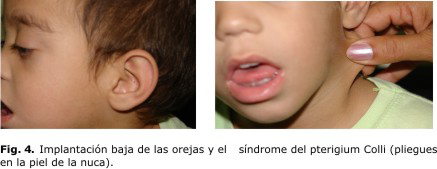
Se observaron otras alteraciones (fig. 4) como la implantación baja de las orejas y el síndrome del Pterigium Colli (pliegues en la piel de la nuca).
El varón presentaba paladar ojival (paladar en forma de bóveda) y aspecto típico de la cara con filtrum (surco vertical en el centro del labio superior) marcado (fig. 5).

Exámenes complementarios de ambos casos
- Ecocardiograma: Compatible con una estenosis pulmonar grado 1 en la hembra y en el varón estenosis valvular con gradiente de 21 milímetros.
- Ultrasonido abdominal: Agenesia vesicular con esteatosis hepática en la niña.
- Talla para la edad: Percentil entre el 3 y 10 en el caso de la niña.
CONCLUSIONES
Todo paciente que presente un síndrome de origen genético que se asocie a alteraciones oftalmológicas debe ser valorado por la consulta de Oftalmología Pediátrica y Estrabismo del Instituto Cubano de Oftalmología "Ramón Pando Ferrer".
REFERENCIAS BIBLIOGRÁFICAS
1. Fundación ONCE - Fondo Europeo de Desarrollo Regional [pagina de Internet]. Madrid: Discapnet; 2006 [citado: 15 ene 2008]. Disponible en: http://salud.discapnet.es/Castellano/Salud/Enfermedades/ EnfermedadesDiscapacitantes/S/Sindrome%20de%20Noonan/Paginas/cover%20noonan.aspx
2. Noonan JA, Ehmke DA. Associated noncardiac malformations in children with congenital heart disease. J Pediatr. 1963;31:150-3.
3. DermAtlas.org [homepage on Internet] Johns HopKins University; 2000-2008 [citado: 18 mayo 2008]. Disponible en: http://dermatlas.med.jhmi.edu/derm/
4. Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukaemia. Blood. 2004;104:307-13.
5. Jongmans M, Sisterman EA, Rikken A, Nillesen WM, Tamminga R, Patton M, et al. Genotypic and phenotypic characterizacion of Noonan syndrome: New data and review of the literature. Am J Med Genet. 2005;134:165-70.
6. Van de Burgt I, Berends E, Lommen E, Van Beersum S, Hamel B, Mariman E. Clinical and molecular studies in a large Dutch family with Noonan syndrome. Am J Med Genet. 1994;53:187-91.
7. Aracena AM. Cardiopatías congénitas y síndromes malformativos-genéticos. Rev Chil Pediatr [serie en Internet]. 2003 [citado: 3 septiembre de 2007];74(4):[aprox. 2 p.]. Disponible en: http://es.wikipedia.org/wiki/S%C3%ADndrome_de_Noonan
Recibido: 21 de enero de 2008.
Aprobado: 3 de marzo de 2008.
Dra. Lucy Pons Castro. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave 76 No. 3104 entre 31 y 41 Marianao, Ciudad de La Habana, Cuba. E-mail: lucypons@infomed.sld.cu

