










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión impresa ISSN 0864-2176versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.22 n.2 Ciudad de la Habana jul.-dic. 2009
PRESENTACIÓN DE CASOS
Síndrome de Weill-Marchesani
Weill-Marchesani syndrome
Seydel Legra NápolesI; Rosa Maria Naranjo FernándezII; Lucy Pons CastroI; Teresita de J. Méndez SánchezII; Mirta Silveira SimónI
IEspecialista de I Grado en Oftalmología. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
IIEspecialista de II Grado en Oftalmología. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
RESUMEN
El síndrome de Weill-Marchesani es un desorden genético poco frecuente del tejido conectivo con afectación ocular. Desde su descripción por Weill y Marchesani en 1932 y 1939, se han descrito patrones de herencia autosómica dominante y recesiva. En general estos pacientes se caracterizan por baja talla, braquidactilia con rigidez articular, microsferofaquia, miopía lenticular progresiva, luxación cristaliniana, y glaucoma secundario. Se presentan las características oftalmológicas y clínicas de una paciente a quien se le diagnosticó este síndrome genético. Procedía de una familia de 4 miembros donde uno de ellos presentaba similares características (padre), no se detectaron malformaciones cardiovasculares asociadas pero se recogen antecedentes de autoagresión. El desempeño del oftalmólogo en su diagnóstico precoz y manejo, es de vital importancia, de esta forma se podría lograr una rehabilitación visual y la consecuente incorporación a una vida socialmente útil.
Palabras clave: Síndrome de Weill Marchesani /etiología; síndrome de Weill Marchesani /genética; anomalías múltiples; sistémicas y oftalmológicas.
ABSTRACT
Weil Marchesani syndrome is a rare genetic disorder of the connective tissue with ocular effect. Since the description of this disease by Weill and Marchesani in 1932 and 1939 respectively, patterns of autosomal dominant and recessive inheritance have been outlined. In general, these patients are characterized by small size, brachydactilia, joint rigidity, microspherophakia, progressive lenticular myopia, crystalline luxation and secondary glaucoma. This paper presented the ophthalmologic and clinical characteristics of a female patient who was diagnosed with this genetic syndrome. She came from a four-member family in which one of them presented with similar characteristics (father); there were not associated cardiovascular malformations, but self-attack history was included. The ophthalmologist's performance in the early diagnosis and management of the disease is of vital importance, because in this way, visual rehabilitation could be materialized, with subsequent incorporation to socially useful life.
Key words: Weill Marchesani syndrome/etiology; Weil Marchesani syndrome/genetics; multiple anomalies; systemic and ophthalmologic anomalies.
INTRODUCCIÓN
El síndrome de la braquidactilia_esferofaquia fue descrito originalmente por Weill en 1932 y fue asociado por este autor con el síndrome de Marfan.1 En 1939 Marchesani separa ambos síndromes, completó así la descripción de Weill y clasificó estos cuadros como distrofias congénitas del mesodermo. Marchesani los divide en: formas hipoplásica (Marfan) e hiperplásica (W. Marchesani). Su frecuencia es bastante baja, son pocos los casos publicados en la literatura mundial, se plantea que 1 de cada 100 000 niños puede presentarlo. Se ha sugerido el acrónimo inglés GEMSS (glaucoma, ectopia lentis, microsferofaquia, rigidez articular, baja estatura) para describir la forma dominante.
Los signos más relevantes están dados por talla baja, cráneo ancho, órbitas pequeñas y poco profundas, hipoplasia maxilar paladar estrecho, mala implantación dentaria, braquidactilia con metacarpianos y falanges anchos y cortos, puede presentar cardiopatía: comunicación interventricular (CIV).2 En los ojos el cristalino es pequeño, esférico, con miopía lenticular y un glaucoma. Un tercio de ellos puede llegar a la ceguera.3
El diagnóstico clínico del síndrome de Weill-Marchesani se basa en las anomalías cristalinianas y esqueléticas. Actualmente se considera que la esferofaquia no es imprescindible para el diagnóstico.1 Otros hallazgos menos frecuentes son la licuefacción vítrea, degeneración coriorretiniana, megalocórnea y microcórnea.
La mayor parte de los cambios fisiológicos cristalinianos ocurren en la superficie anterior del cristalino, el cual se torna más esférico a medida que envejece.2
La causa de la microsferofaquia es desconocida, aunque se ha propuesto que sea debida a una alteración en el desarrollo de las fibras cristalinianas secundarias y/o a un defecto en la inserción de las fibras secundarias anómalamente finas.4 Otras teorías atribuyen la falta de tensión en sus fibras zonulares rudimentarias, impidiendo el desarrollo fisiológico del cristalino y ocasionando su forma esférica. Una anomalía en las fibras zonulares permitiría al cristalino permanecer esférico en lugar de tender a una forma biconvexa con el paso de los años.1 Todo esto, ligado a la tendencia de la luxación y del desplazamiento del cristalino explicarían la progresión de la miopía lenticular. La pérdida de visión obedece a la miopía. La miopía significativa aparece a la edad media de 11,2 años, la subluxación cristaliniana sucede a los 18,2 años; su desplazamiento suele ocurrir inferiormente.
Las indicaciones para la cirugía del cristalino: son su dislocación a cámara anterior, subluxación progresiva con bisección pupilar, dislocación inminente, diplopía monocular y agudeza visual menor de 20/70 para lejos y de 15 para cerca.2
El manejo actual comprende la extracción cristaliniana con o sin colocación de lente intraocular y vitrectomía anterior,4 además del tratamiento médico o quirúrgico del glaucoma secundario. No obstante, existe un mayor riesgo de complicaciones posquirúrgicas en estos pacientes, como atalamia tras cirugía filtrante5 o roturas zonulares.2-5
La corrección óptica precoz previene la ambliopía, y el seguimiento del paciente permite detectar y tratar complicaciones oftalmológicas, como es el glaucoma secundario.
PRESENTACIÓN DEL CASO
Paciente femenina de 5 años de edad, raza blanca, quien al nacer sus padres le detectaron malformaciones en la oreja y en los miembros superiores (manos) con bifurcación del pulgar derecho, braquimorfia y braquidactilia en las cuatro extremidades. Fenotípicamente su talla es corta con una estatura de 102 cm, peso 18 kg. Durante su crecimiento y desarrollo observaron su mala visión (acerca los objetos para definirlos). A los 4 años de edad inicia las actividades escolares y es más notable el empeoramiento de la visión, esta se acompañaba de cefalea frontal que mejora con el reposo visual, por lo cual fue valorada en el Servicio de Oftalmología Pediátrica, donde se le diagnosticó defectos de refracción elevados en ambos ojos, (miopía) y (astigmatismo), epicantus, córnea pequeña, cámara anterior estrecha, presiones intraoculares normales, esferofaquia y subluxación del cristalino inferior, además de un síndrome genético.
Las imágenes que se presentan a continuación fueron obtenidas y se publican con el consentimiento del paciente y sus familiares.
Exámenes realizados
Agudeza visual sin corrección (AV sc)
Ojo derecho (OD): 0,05 Ojo izquierdo (OI): 0,1
Presión intraocular (PIO)
OD: 22 mmHg OD: 23 mmHg x aire
Corrección óptica
OD: -5,00 -12,00 x 175 (0,1) OI: -6,00 -13,00 x 170° grado (0,1)
Queratometría
| OD: | 52,25 | OI: | 51,50 |
| 46,50 | 46,00 |
Biometría
OD: LAX 20,5 OI: LAX 21,1
CA: 4,37 CA: 4,69
Paquimetría
OD: 599 OI: 608
Lentes de contacto
OD: 7,40 -2,OO OI: 7,30 -1,00
Se realizó interconsulta con el Servicio de Córnea y Lentes de Contacto; se indicó lentes, y se inició la rehabilitación visual y el seguimiento por baja visión.
Todo paciente que presente alteraciones oftalmológicas asociadas a un síndrome de origen genético, debe ser tratado de forma precoz y continuar su seguimiento para diagnosticar posibles complicaciones que conlleven al deterioro visual y por ende a la ceguera.
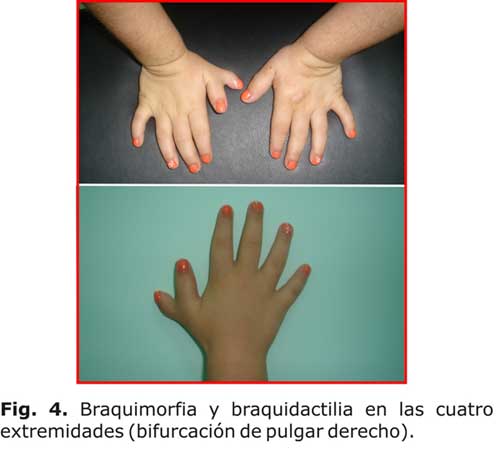
Alteraciones esqueléticas
Talla baja, cráneo ancho, órbitas pequeñas y poco profundas, hipoplasia maxilar paladar estrecho, mala implantación dentaria, braquidactilia con metacarpianos y falanges anchos y cortos, puede presentar cardiopatía: comunicación interventricular (CIV) (fig.1,2,3,4).2


Alteraciones oftalmológicas
Epicantus, córnea pequeña, cámara anterior estrecha, dislocación del cristalino a cámara anterior con glaucoma secundario, esferofaquia y microsferofaquia, miopía lenticular progresiva, luxación cristaliniana, y/o desplazamiento del cristalino lo que explicaría la progresión de la miopía lenticular, subluxación progresiva con bisección pupilar, dislocación inminente, diplopía monocular y agudeza visual menor de 20/70 para lejos y de 15 para cerca (fig. 5).
REFERENCIAS BIBLIOGRÁFICAS
1. Veiga de la Jara C, Bosch Valero J, Torres Suárez E, Mateos Sánchez E, Rojo Castejón P, Ancochea Díaz G. Síndrome de Weill Marchesani: afectación familiar. Arch Soc Esp Oftalmol. [Seriado en Internet]. 2006 [citado 12 abril 2009]; 81(6): [aprox. 4 p.]. Disponible en: http://scielo.isciii.es/pdf/aseo/v81n6/comunicacion5.pdf
2. Herrera J, Morales M. Síndrome de Weill—Marchesani. Rev Chile Ped. 1986; 57(6):571-572.
3. Evereklioglu C, Hepsen IF, Er H. Weill-Marchesani syndrome in three generations. Eye. 1999;13:773-7.
4. Wirtz MK, Samples JR, Kramer PL, Rust K, Yount J, Acott TS, et al. Weill-Marchesani syndrome-possible linkage of the autosomal dominant form to 1521.1. Am J Med Genet. 1996;65:68-75.
5. Reza Razeghinejad M, Safavian H. Central Corneal Thickness in Patients With Weill-Marchesani Síndrome. Am J Ophthalmol. [Seriado en intenet]. 2006 [citado 15 marzo 2009];142(3):507-8. Disponible en: http://www.ajo.com/article/S0002-9394(06)00434-X/abstract
6. Chu BS. Weill-Marchesani syndrome and secondary glaucoma associated with ectopia lentis. Clin Exp Optom. 2006;89(2):95-9.
7. Weill. G. Ectopie des cristallins et malformations générales. Annales d'oculistique. 1932;169:21-44.
8. Marchesani O. Brachydaktylie und angeborene Kugellinse als Systemerkrankung. Klinische Monatsblätter für Augenheilkunde. Stuttgart. 1939;103:392-406.
9. Kloepfer HW, Rosenthal SW. Possible genetic Carriers in the spherophakia-brachymorphia syndrome. Am J Hum Genet. 1955;7:398-425.
10. McGavic JS. Weill-Marchesani syndrome: brachymorphism and ectopia lentis. American Journal of Ophthalmology. 1966;62:820-3.
Recibido: 20 de marzo de 2008.
Aprobado: 15 de julio de 2008.
Dra. Seydel Legra Nápoles. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave. 76 No. 3104 entre 31 y 41 Marianao, Ciudad de La Habana, Cuba. E-mail: seydi@jagua.cfg.sld.cu


{kind=link}
{kind=link}