










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.23 supl.1 Ciudad de la Habana 2010
PRESENTACIONES DE CASOS
Apraxia oculomotora congénita
Congenital oculomoror apraxia
Rosa M. Naranjo FernándezI; Pedra RodríguezII; Yaimir Estévez MirandaIII; Teresita de J. Méndez SánchezIV
IEspecialista de II Grado en Oftalmología. Asistente. Instituto Cubano de Oftalmología. «Ramón Pando Ferrer». La Habana, Cuba.
IIEspecialista de II Grado en Oftalmología. Profesor Auxiliar. Cenrtro Provincial de Retinosis Pigmentaria. Sancti Spiritus, Cuba.
IIIEspecialista de I Grado en Oftalmología. Especialista de I Grado en Medicina General Integral. Instructor. Instituto Cubano de Oftalmología «Ramón Pando Ferrer». La Habana, Cuba.
IVEspecialista de I Grado en Oftalmología. Instructor. Instituto Cubano de Oftalmología. «Ramón Pando Ferrer». La Habana, Cuba.
RESUMEN
La apraxia oculomotora congénita, también conocida como Síndrome de Cogan, es una enfermedad hereditaria rara que afecta al ojo, caracterizada por la incapacidad para ejecutar movimientos oculares horizontales voluntarios. Descrita por Cogan en 1952, de etiología desconocida, clásicamente se le considera un desorden esporádico, con herencia autosómica dominante en algunos casos y en otros sugieren alteraciones metabólicas parciales o defectos del desarrollo neurológico. Acude al servicio de oftalmología pediátrica un paciente masculino de ocho meses de edad y al examen de motilidad ocular se detecta que cuando el niño intenta mover los ojos hacia el objeto que le llama la atención, realiza movimientos bruscos laterales de la cabeza. Se realizan estudios de neuro imágenes y electroencefalograma, no se encontraron alteraciones. Este es un diagnóstico de exclusión donde deben descartarse otras causas de defecto de fijación y movimientos cefálicos anómalos. En este momento con dos años de edad se constata una mejoría dada por disminución de las sacudidas cefálicas. Sin embargo, el retraso del desarrollo psicomotor, especialmente del lenguaje, sí puede requerir educación especial. Esta enfermedad es poco frecuente por lo que su detección temprana contribuye a un mejor pronóstico visual.
Palabras clave: Apraxia oculomotora, congénito, Síndrome de Cogan, niño.
ABSTRACT
The congenital ocular motor apraxia, also known as Cogan´s Syndrome, is a rare heredity disease that affects the eye in that to the extent that it can not move horizontally at will. Of unknown etiology, it was described by Cogan in 1952 and, classically considered as a sporadic disease with dominant autosomal heredity in some cases, and as an indicator of partial metabolic alterations or neurological development defects in others. An 8 month-old male patient was seen at the pediatric ophthalmology department. On the ocular motility exam, it was found that the child attempted to move his eyes towards an object that got his attention, but instead of this, he moved his head laterally and abruptly. Some studies based on neuroimaging and electroencephalograms were performed; No alteration was found. This is an exclusion diagnosis because other causes of fixation defects and anomalous head movements should be ruled out. Presently, the patient is 2 years old and has improved due to decrease in sudden head movements. However, his retarded psychomotor development, particularly speaking, does require special education. This disease is not common, that is why early detection may contribute to a better visual prognosis.
Key words: Ocular motor apraxia, congenital, Cogan syndrome, child.
INTRODUCCIÓN
La apraxia oculomotora congénita, también conocida como Síndrome de Cogan, es una enfermedad hereditaria rara que afecta al ojo y está presente desde el nacimiento. Es una alteración de la función cerebral caracterizada por la incapacidad para ejecutar movimientos oculares horizontales voluntarios.1
Es un síndrome poco frecuente,2 descrito por Cogan en 1952 y desde entonces han sido reportados varios casos. En algunos se han encontrado anomalías sistémicas o herencia familiar, sin embargo, la mayoría de son esporádicos sin anomalías sistémicas.
Es una enfermedad de etiología desconocida, clásicamente se le considera un desorden esporádico, aunque se han comentado algunos casos con herencia autosómica dominante. Otras publicaciones sugieren que puede deberse a alteraciones metabólicas parciales o defectos del desarrollo neurológico, como hipoplasia del vérmix cerebeloso, aplasia del cuerpo calloso, heterotopias de la sustancia gris, síndrome de Kallman, macrocerebelo y alteraciones cromosómicas,3 pero sin haberse demostrado asociación constante.1
Como enfermedades graves asociadas a esta entidad se han descrito la neurofibromatosis, la degeneración olivo-ponto-cerebelosa, la corea de Huntington, el síndrome de Coach, la enfermedad de Gaucher, ataxia-telangiectasia y ataxia-apraxia oculomotora.1
Los movimientos sacádicos, rápidos, dirigidos a cambiar la fijación de la mirada, aparecen unas semanas tras el nacimiento y son controlados supranuclearmente por los lóbulos frontales (las sacudidas horizontales por el lóbulo frontal contralateral; las verticales de forma bilateral por ambos lóbulos frontales). Los movimientos de persecución son aquellos automáticos y lentos que permiten seguir un objeto que se desplaza. Estos dependen de la región parieto-occipital ipsilateral. Los reflejos vestíbulo-ocular y optocinético comparten en su fase rápida la vía de los movimientos sacádicos.2
Clínicamente deriva de la incapacidad de refijación sacádica en el plano horizontal, el paciente no puede cambiar voluntariamente la dirección de su mirada hacia los laterales ni presenta la fase rápida de los reflejos vestíbulo-ocular ni optocinético. Para compensar ese déficit y conseguir refijar la mirada, desarrolla las características sacudidas cefálicas laterales: el niño gira la cabeza rápidamente hacia el nuevo punto de interés, sobrepasándolo. La fase lenta del reflejo óculo-vestibular lleva los ojos en la dirección opuesta a la del movimiento cefálico; los ojos quedan orientados hacia el objeto deseado y se consigue la refijación.2,4. Lentamente la cabeza vuelve a su posición centrada sin alterar la nueva dirección de fijación. Las sacudidas suelen iniciarse a los 4-8 meses de edad.2
La forma congénita de esta alteración se presenta con más frecuencia en niños, por lo general normales, aunque también se ha visto asociada a defectos del sistema nervioso central que incluyen degeneraciones espinocerebelosas, retardo mental, hidrocefalia y tumores de la fosa posterior o del tallo cerebral.5
La apraxia oculomotora adquirida, también conocida como Síndrome de Balint, se caracteriza por parálisis de los movimientos sacádicos y de seguimiento voluntarios en todas las direcciones de mirada y ausencia del nistagmo optocinético (NOC). El nistagmus vestibular y los movimientos oculares causales están intactos. Se asocia con demencia y alteraciones del campo visual. Ha sido descrita su relación con la encefalitis y lesiones del lóbulo parietal.5
La observación del comportamiento del paciente frente al examen de motilidad ocular, ducciones y versiones es fundamental. Ante la sospecha de esta entidad, debe dejarse al paciente mover su cabeza libremente y luego, sosteniéndola, asegurarse de la perfecta excursión de los músculos extraoculares. El movimiento de cabeza es característico y por ello distinguible de otros movimientos de cabeza o posiciones compensadoras presente en paresias o parálisis de los nervios craneanos, nistagmus, spasmus nutans, etc. Cuando el bebé es muy pequeño, los potenciales visuales evocados, que son normales, pueden ser de gran ayuda diagnóstica.
PRESENTACIÓN DEL CASO
Paciente masculino de 8 meses de edad atendido en el Servicio de Oftalmología Pediátrica del Instituto Cubano de Oftalmología «Ramón Pando Ferrer». La madre refiere que el paciente no fija la vista. No hay información de antecedentes heredofamiliares ni personales patológicos.
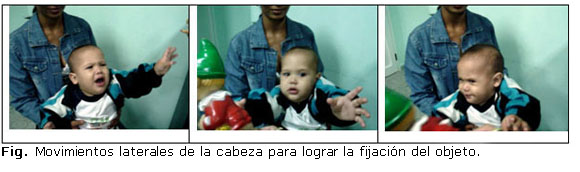
En el examen oftalmológico de ambos ojos se encontró: anexos y segmento anterior normal, medios transparente y fondo de ojo sin alteraciones. Al explorar la motilidad ocular se detecta que cuando el niño intenta mover los ojos hacia el objeto que le llama la atención, realiza movimientos bruscos laterales de la cabeza ( figura ). En la esquiascopia se corrige para ambos ojos con +2,0 Dioptrías de esfera.
Por todo lo anterior la impresión diagnóstica fue apraxia oculomotora congénita. Se decide realizar una interconsulta para que el paciente sea valorado por el servicio de Neuroftalmología en nuestro centro y por el Instituto de Neurología y es confirmado el diagnóstico. En los estudios realizados de neuroimágenes y en el electroencefalograma (EEG) no se encontraron alteraciones.
En la actualidad el paciente tiene 2 años de edad y se constata una mejoría, con disminución de las sacudidas cefálicas y de la capacidad de refijación.
DISCUSIÓN
La apraxia oculomotora congénita es una ausencia congénita de la mirada lateral conjugada, compensada mediante un giro violento de la cabeza en el sentido deseado para contrarrestar el desplazamiento pasivo que sufren los globos oculares en sentido opuesto. Posteriormente, la cabeza retorna a su posición primitiva.
En algunos casos se asocia con otras anomalías, la mayor parte de los pacientes son casos aislados. Una característica muy importante en esta entidad es la aparición de movimientos cefálicos compensatorios, los cuales se van haciendo menos evidentes al crecer el paciente.1
Esta enfermedad se diagnostica cuando hay una serie de datos clínicos oculares y de movimientos cefálicos que se desarrollan durante el primer año de vida. Los niños afectados parecen no tener fijación visual por lo que muchas veces se cree que pueden ser ciegos. Cuando existen signos de disfunción neurológica aparecen de manera temprana.
La apraxia oculomotora congénita debe ser un diagnóstico de exclusión, debe descartarse otras causas de defecto de fijación y movimientos cefálicos anómalos: déficit visual por problema ocular (cataratas, transtornos de polo posterior),2 alteraciones de la movilidad ocular (ej. síndrome de Duane)1 o neuroftalmológico (atrofias ópticas, lesiones en SNC) y procesos que cursen con movimientos cefálicos anómalos (epilepsias de aparición temprana como el síndrome de West, caracterizado por espasmos, retraso psicomotor y alteraciones electroencefalográficas)2, o incluso sospechar alteraciones psicológicas que nos enfrenten a un simulador. Se desconoce la causa de este trastorno. Hay varios reportes acerca de casos con defectos del desarrollo neurológico, pero son considerados esporádicos.1
Para establecer el diagnóstico es necesario comprobar que se conservan los movimientos sacádicos verticales (tanto voluntarios como en los reflejos oculo-vestibular y optocinético). En la apraxia oculomotora adquirida (secundaria a lesiones adquiridas del SNC (tumores de fosa posterior, lesiones frontoparietales bilaterales) también se afectan los verticales.2
Ante un paciente con déficit de abducción bilateral, los principales diagnósticos diferenciales a tomar en cuenta son: síndrome de Duane tipo I bilateral, parálisis de nervio abducens bilateral, síndrome de Möbius y endotropía infantil o congénita.2
Cuando hemos confirmado el diagnóstico, se debe tranquilizar a los padres ya que, con el tiempo el cuadro mejora en casi todos los casos, disminuye el movimiento cefálico compensatorio y mejora la capacidad de refijación. Sin embargo, el retraso del desarrollo psicomotor, especialmente del lenguaje, sí puede requerir educación especial.
Es importante publicar este caso ya que la apraxia oculomotora congénita es una entidad rara, poco frecuente y es necesario su conocimiento. De esta forma podemos diagnosticarla en el momento oportuno y corregir el defecto a una temprana edad.
REFERENCIAS BIBLIOGRÁFICAS
1. Gulias-Cañizo R, Sánchez-Huerta V, Rubio-Lezama M. Apraxia oculomotora congénita: informe de un caso. Rev Mex Oftalmol. 2005;79(6):341-3.
2. De Benito L, Merino P, Gómez de Liaño P, Franco G, Herrera J. Consideraciones sobre el síndrome de Cogan (apraxia oculomotora congénita) a partir de un caso. Arch Soc Esp de Oftalmol. 2004;79(4):189-92.
3. Betz R, Rensing C, Otto E, Mincheva A, Zehnder D, Lichter P. et al. Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr. 2000;136:828-31.
4. Federación Española de Asociaciones de Enfermedades Raras. [Página principal en Internet]. [citado 7 de dic. de 2009]. Apraxia Oculomotora de Cogan. Disponible en : http://www.enfermedades-raras.org/es/default.htm
5. Acosta Ureña V. Apraxia oculomotor. Franja Visual. 1996;7(27):31-2.
Recibido: 26 de noviembre de 2010
Aprobado: 5 de diciembre de 2010
Dra. Rosa M. Naranjo Fernández. Instituto Cubano de Oftalmología «Ramón Pando Ferrer». Ave. 76 No. 3104 entre 31 y 41 Marianao. La Habana, Cuba. Correo electrónico: rnfernandez@infomed.sld.cu


{kind=link}