










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión impresa ISSN 0864-2176
Rev Cubana Oftalmol vol.25 supl.1 Ciudad de la Habana 2012
PRESENTACIONES DE CASOS
Síndrome de Axenfeld-Rieger con glaucoma asociado
Axenfeld-Rieger syndrome associated with glaucoma
Dra. Yanileidy Blanco González, Dra. Teresita de Jesús Méndez Sánchez, Dra. Liamet Fernández Argones, Dr. Daniel López Felipe, Dr. Ibraín Piloto Díaz, Dra. Lourdes Rita Hernández Santos
Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
RESUMEN
Se describen dos casos clínicos con síndrome de Axenfeld-Rieger y glaucoma bilateral. El primer caso representa una paciente femenina con corectopia, embriotoxon posterior, atrofia del estroma anterior del iris, adherencias iridocorneales y glaucoma bilateral; por lo que se decide realizar trabéculo-trabeculectomía en ambos ojos y mantuvo cifras de tensión intraocular dentro de límites normales al año de evolución. El segundo caso se trata de un paciente masculino con escleras azules, microcórnea, embriotoxon posterior, aniridia parcial, atrofia sectorial del iris y glaucoma bilateral; por lo que se realizó trabéculo-trabeculectomía de ambos ojos con descompensación posterior del ojo izquierdo. Se inició el tratamiento médico sin buenos resultados y se reintervino el ojo izquierdo, se descompensó dos meses después de la cirugía. Actualmente se encuentra compensado con tratamiento médico.
Palabras clave: síndrome de Axenfeld-Rieger, glaucoma congénito, niño.
ABSTRACT
Two clinical cases of Syndrome of Axenfeld–Riegel and bilateral glaucoma were described. The first case was a female patient with corectopia, posterior embriotoxon, atrophy of the anterior stroma of the iris, iridocorneal adherences and bilateral glaucoma. To treat her, it was decided to perform trabeculo-traceculectomy in both eyes and as a result, the patient kept intraocular pressure values within the normal limits after one year. The second case was male patient who suffered with blue sclera, microcornea, posterior embrotoxon, partial aniridia, sector atrophies of the iris and bilateral glaucoma. Trabeculo-trabeculectomy of both eyes with posterior decompensation of the left eye was applied. The medical treatment did not yield good results, His left eye was then re-operated and two month afterwards, decompensation occurred again. At present, he is compensated with a new medical treatment.
Key words: Axenfeld-Rieger’s syndrome, congenital glaucoma, child.
INTRODUCCIÓN
El síndrome de Axenfeld-Rieger (SAR) es una entidad rara, autosómica dominante, que se caracteriza por una penetrancia completa y expresividad variable.1 Los defectos oculares clásicos del síndrome de Axenfeld-Rieger incluyen hipoplasia del iris, adherencias iridocorneales, corectopia, policoria, embriotoxon posterior, y otros rasgos menos frecuentes como cataratas, desprendimiento de retina y microcórnea.2,3 Las anomalías sistémicas asociadas incluyen las malformaciones faciales (telecantus, hipoplasia del maxilar), anomalías dentarias y piel periumbilical redundante entre otras. Aproximadamente la mitad de los individuos afectados desarrollan glaucoma.4,5
Se describen dos casos clínicos con el objetivo de mostrar lo interesante de la asociación del síndrome de Axenfeld-Rieger y el glaucoma en la edad pediátrica. También se analiza la evolución, manejo y pronóstico visual de estos pacientes.
PRESENTACIONES DE CASOS
Caso 1
Paciente de 1 año de edad, femenina, con diagnóstico de síndrome de Axenfeld-Rieger. Fue estudiada por genética y se detectó que el padre mantiene seguimiento por glaucoma asociado a este síndrome.
Al examen oftalmológico se apreció hipertelorismo y buftalmo en ambos ojos (AO). En el segmento anterior de AO se observó megalocórnea, embriotoxon posterior, adherencias iridocorneales, corectopia, atrofia del estroma anterior del iris y ectropión uveal. Los medios refringentes muestran un edema corneal ligero (AO) que permite la visualización del cristalino subluxado en el ojo izquierdo (OI) (Fig. 1).

En el examen bajo anestesia se encontró a la fundoscopia una excavación de 0,4 ojo derecho (OD) y 0,7 OI con disminución del anillo neurorretiniano en sector temporal de este último ojo. Los valores del diámetro corneal horizontal fueron en el OD de 13 mm y en el OI de 13,5 mm; el diámetro vertical del OD 11 mm y del OI 11,5 mm. La longitud axial medida por biometría mostró valores de 22,46 mm y 22,71 mm en el OD y OI respectivamente, con presiones intraoculares con tonómetro de Perkins de 24 mmHg en OD y en el OI de 28 mmHg.
A la gonioscopia se observó una línea de Schwalbe prominente desplazada en sentido anterior, inserción anterior del iris en sector inferior y superior, y procesos iridianos que llegan hasta la línea de Schwalbe en sector temporal y nasal. En el ultrasonido no se encontró alteración del polo posterior.
Dentro de las alteraciones no oftalmológicas se apreció un puente nasal ancho, hipoplasia del maxilar inferior, alteraciones dentarias (microdontia), pliegue umbilical redundante y dilatación del III ventrículo.
Por los antecedentes familiares, los hallazgos encontrados al examen físico y el estudio genético realizado se llegó al diagnóstico de síndrome de Axenfeld-Rieger con glaucoma bilateral asociado. Se decidió realizar trabeculo-trabeculectomía en ambos ojos.
En la exploración bajo anestesia a los 3 meses de la cirugía se constataron córneas transparentes, disminución de la excavación del OD a 0,3 y valores de presión intraocular (PIO) de 9 mmHg en OD y 10 mmHg en OI. No hubo variación en los valores del diámetro corneal y la biometría. En las consultas de seguimiento trimestrales ha mantenido valores normales de PIO.
Caso 2
Paciente de 9 años de edad, masculino, con diagnóstico de síndrome de Axenfeld-Rieger. Se estudió por genética, fue operado de oclusión intestinal y reconstrucción umbilical, sin antecedentes familiares de enfermedad ocular y madre con hipotiroidismo.
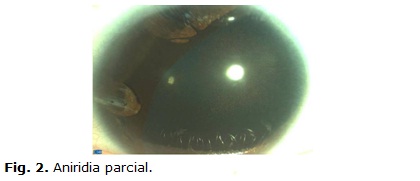
La agudeza visual sin cristales fue de 0,1 OD y 0,2 OI y la mejor corregida de 0,6 OD y 0,7 OI. Presentaba un defecto refractivo de -2,50 -0,50 x 65° en OD y de -4,00 -2,50 x 25° en el otro ojo. Al examen oftalmológico de AO se encontró escleras azules. En el segmento anterior se observó microcórnea, embriotoxon posterior, aniridia parcial, atrofia sectorial del iris (Fig. 2). Los medios refringentes no mostraron edema corneal, solo la presencia de pigmentos a nivel del cristalino de forma bilateral. En el examen del fondo de ojo se halló papilas con excavación de 0,6 en OD y 0,7 en OI, con disminución del anillo neurorretiniano en sector inferior y temporal, y cambios del epitelio pigmentario retinal a nivel ecuatorial.
Los valores del diámetro corneal fueron de 11 mm horizontal y 10,5 mm vertical en ambos ojos. La longitud axial mediante biometría mostró valores de 25,02 mm y 25,11 mm en OD y OI respectivamente. Los valores de presión intraocular (PIO) con tonómetro de Pascal fueron de 40 mmHg en OD y 39 mmHg en el otro. Los valores de paquimetría central fueron de 677 y 655 mm en cada ojo.
Al examen por gonioscopia se observó la línea de Schwalbe prominente y desplazada anteriormente, inserción anterior del iris en sector inferior y superior, procesos iridianos que llegan hasta el trabéculo pigmentado. Al ultrasonido no se encontraron alteraciones. En los estudios de campo visual automatizado umbral se mostró un aumento de la mancha ciega hacia superior en OD y en OI un defecto arciforme superior.
Dentro de las alteraciones no oftalmológicas se encontró hipoplasia del maxilar inferior, alteraciones dentarias, pliegue umbilical redundante y alteraciones osteomioarticulares. (Fig. 3).

Por los hallazgos encontrados al examen físico y el estudio genético realizado se llegó al diagnóstico de síndrome de Axenfeld-Rieger con glaucoma bilateral asociado. Se realizó trabéculo-trabeculectomía en los dos ojos.
En el posoperatorio inmediato la PIO fue de 15 mmHg en OD y 17 mmHg en OI con tonómetro de Pascal. Las paquimetrías de 620 mm OD y 630 mm OI con reducción del grosor corneal de 57 mm y 25 mm respectivamente.
Al mes los valores de la PIO alcanzaron 18 mmHg en OD y 31 mmHg en OI medidos con tonómetro de Pascal. Entonces se introduce tratamiento con timolol 0,5 % en el ojo izquierdo. No se encontraron variaciones evolutivas en el examen de fondo de ojo.
Al no lograrse reducción de las presiones intraoculares en el OI, se decidió realizar trabéculo-trabeculectomía, que fue efectiva durante 2 meses volviéndose a indicar tratamiento médico con timolol 0,5 %. Se ha mantenido compensado hasta el momento con cifras de PIO de 10 mmHg en OD y 17 mmHg en OI por tonometría de Pascal.
DISCUSIÓN
El síndrome de Axenfeld-Rieger se considera una enfermedad ocular congénita dentro de las alteraciones del desarrollo que involucran a las estructuras angulares. Estos cambios morfológicos conducen a su asociación con el glaucoma. Frecuentemente las alteraciones oculares coexisten con otros hallazgos sistémicos que caracterizan el síndrome.6
En la anomalía de Axenfeld solo se observa embriotoxon y trastornos en el ángulo. La anomalía de Rieger presenta las alteraciones del Axenfeld y se suman otras en el iris. El síndrome de Rieger tiene, además del embriotoxon, compromiso del seno camerular, del iris y alteraciones sistémicas. Estos cambios oculares son los que permiten diferenciar las distintas anomalías, del síndrome de Axenfeld-Rieger. Excepto cuando la única alteración que se encuentra es el embriotoxon posterior, el resto de manifestaciones tiene riesgo de desarrollar glaucoma.7
La transmisión es usualmente dominante (75 %) para el grupo de Axenfeld-Rieger, pero puede ser esporádico.8 Se han identificado 5 sitios cromosómicos (4q25, 6p25, 11p13, 13q14, 16q24) y 3 genes de factor de transcripción. Dos de estos genes, PITX2 y FOXC1, que se vinculan a los cromosomas 4q25 y 6p25 respectivamente, son los más frecuentemente afectados. 9
Los hallazgos clínicos pueden ser divididos en oculares y no oculares (sistémico). Las alteraciones oculares observadas afectan principalmente al iris, córnea y cámara anterior.
Los cambios a nivel del iris son de gran importancia al establecer el diagnóstico del SAR, e incluyen hipoplasia, corectopia y policoria. Los trastornos iridianos pueden ser muy sutiles y parecer un iris normal, sin el examen del ángulo iridocorneal. En el primer caso descrito se apreció corectopia y atrofia del estroma anterior del iris. El segundo paciente presentó aniridia parcial bilateral y atrofia sectorial del iris.
La línea de Schwalbe es prominente y se sitúa anteriormente (embriotoxon posterior). Aparece como una línea blanca en la córnea posterior cerca del limbo y puede observarse en lámpara de hendidura o en la gonioscopia. El embriotoxon posterior se encuentra en la mayoría de los pacientes, pero aproximadamente 15 % de la población general lo presenta, sin riesgo de desarrollar glaucoma. Cuando se identifica en un paciente con disgenesia del segmento anterior, la primera consideración debe ser síndrome de Axenfeld-Rieger. La ausencia de otras alteraciones corneales como megalocórnea, esclerocórnea u opacidades corneales, son criterios para diferenciar el SAR de otras disgenesias del segmento anterior.10
Gonioscópicamente se aprecia el embriotoxon posterior asociado a adhesiones iridocorneales. Estas son tiras de tejido similar en color y textura al iris, que comunican el ángulo camerular desde el iris periférico hasta la prominencia formada por la línea de Schwalbe. Por detrás de ellas, se encuentra un ángulo abierto, con el trabéculo visible y el espolón escleral parcialmente oculto por una inserción iridiana en la porción posterior de la red trabecular (inserción alta del iris).6
El glaucoma es el problema ocular más serio en estos pacientes, frecuentemente bilateral, se encuentra presente en 50 % de los casos y a veces desde el nacimiento. Puede aparecer durante la niñez, pero es más común durante la adolescencia o al principio de la madurez. Se caracteriza por ser de difícil manejo y tener un pronóstico visual reservado.11
Son posibles otros trastornos oculares: catarata, degeneración macular, hipoplasia del nervio óptico, desprendimiento de retina e hipermetropía. El caso 1 presentó subluxación del cristalino unilateral y el caso 2 pigmentos iridianos a nivel del cristalino, además de cambios del epitelio pigmentario retinal a nivel ecuatorial.
Dentro de las manifestaciones sistémicas se describen las anomalías faciales como la hipoplasia maxilar y mandibular, aplanamiento de la base de la nariz, telecantus e hipertelorismo. Dentro de las anomalías dentarias se encuentran la microdontia, anodontia, oligodontia (predominantes en los incisivos superiores). La piel periumbilical es redundante, presente en los dos casos descritos. Las anomalías óseas y articulares están caracterizadas por apófisis clinoides posteriores prominentes, centro pseudoepifisario del metatarso y metacarpo, osificación irregular de la cabeza femoral y de las epífisis distales tibial y femoral, 2do y 5to metatarsos cortos. En el sistema cardiovascular se incluyen los defectos de los tabiques interauriculares e interventriculares. En el sistema nervioso central se puede detectar hipoacusia neurosensorial, hidrocefalia, calcificaciones leptomeningeas , retraso mental leve y en algunos casos, se observa hipospadia en pacientes masculinos.12
Tanto las alteraciones oculares como sistémicas resultan de la detención en la formación de los tejidos derivados de la cresta neural. Esto incluye el endotelio corneal, ángulo de la cámara anterior e iris.13
Se han identificado muchas enfermedades a considerar en el diagnóstico diferencial como la hipoplasia congénita del iris, la displasia oculodentodigital y la anomalía de Peters. Sin embargo, las 2 más importantes son el síndrome irido-córneo-endotelial y la distrofia polimórfica posterior.
El síndrome irido-córneo-endotelial (SICE) es la condición más frecuentemente confundida con el síndrome de Axenfeld-Rieger. En esta afección el defecto fundamental es una alteración del endotelio corneal que lleva a su proliferación anormal sobre las estructuras corneales, el ángulo y la superficie del iris. Los cambios morfológicos presentan similitudes clínicas e histológicas entre las dos entidades, pero el SICE frecuentemente es unilateral, se inicia en adultos jóvenes y no hay historia familiar positiva. En el síndrome irido-córneo-endotelial raramente se ve el embriotoxon posterior.
En la distrofia polimórfica posterior el endotelio corneal adopta características de epitelio, cubre el trabéculo e iris, y ocasiona una obstrucción al flujo de salida del humor acuoso. El endotelio corneal y la membrana de Descemet tienen apariencia, en la lámpara de hendidura, de vesículas en la superficie posterior de la córnea, que se colocan de forma lineal o en grupos. Un pequeño número de pacientes tienen alteraciones angulares, adhesiones iridocorneales, corectopia y ectropión uveal, con glaucoma o sin este. La distrofia polimórfica posterior es congénita, de herencia autosómica dominante y sin predilección por sexo o raza.6
Los casos con glaucoma plantean un difícil reto terapéutico. Con excepción de los pacientes infantiles, debe intentarse la terapia médica antes de cualquier cirugía. Los fármacos de mayor utilidad son los betabloqueadores, agonistas adrenérgicos alfa 2 e inhibidores de la anhidrasa carbónica.
En los pacientes pediátricos la conducta de elección inicial es la cirugía. Se considera la goniotomía o la trabeculotomía como tratamientos con poca efectividad.
La trabeculectomía es el procedimiento quirúrgico de elección y sus resultados son comparables a los alcanzados con otras formas de glaucoma, que comprometen a pacientes en un rango de edad similar. Desafortunadamente como muchos son niños, tienen un pronóstico más reservado respecto a la utilidad de ésta cirugía por la respuesta fibrótica exagerada, característica de su grupo de edad. En los casos clínicos descritos se empleó la trabeéculo-trabeculectomía sin complicaciones transoperatorias. La trabeculoplastia láser no está indicada porque es técnicamente difícil por la presencia de las adhesiones iridocorneales, la inserción alta del iris, y el incremento del riesgo de formación de sinequias anteriores periféricas que empeorarían el cuadro.6
En la actualidad existe autores que consideran los implantes valvulares como la primera elección quirúrgica. Sobre todo en los síndromes iridocorneales como el SAR, por su elevado índice de fracaso mediante cirugía convencional.14
En la rehabilitación visual debe tenerse presente los errores refractivos, principalmente la miopía en aquellos casos que presentan glaucoma en los 3 primeros años de vida para lograr un mejor pronóstico visual. Esto se debe al aumento de la longitud axial secundaria a la elevación de la presión intraocular y la ambliopía consecuente.
En los casos descritos se muestra, que a pesar de los avances en el diagnóstico y tratamiento del glaucoma, aún no existe una terapia efectiva y única para el tratamiento de casos complejos. Se considera de gran importancia resaltar el manejo adecuado de estos pacientes y su evolución médica, por el reservado pronóstico visual que presentan.
REFERENCIAS BIBLIOGRÁFICAS
1. Hjalt TA, Semina EV. Current molecular understanding of Axenfeld-Rieger syndrome. Expert Reviews Molecular Medicine. 2005;7(25):117.
2. Spallone A. Retinal detachment in Axenfeld-Rieger syndrome. Br J Ophthalmol. 1989;73(7):559-62.
3. Kamiñska A, Sokolowska-Oracz A, Pawluczyk-Dyjeciñska M, Szaflik JP. Variability of clinical manifestations in the family with Axenfeld-Rieger syndrome. Klin Oczna. 2007;109(7-9):321-6.
4. Rand R, Damji K, Freedman S, Moroi S, Shafranov G. Shields' Textbook of Glaucoma. 1ra. ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
5. Alward WL. Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol. 2000;130(1):107-15.
6. Ortiz GE, Nova GM. Síndrome de Axenfeld-Rieger con glaucoma bilateral y descompensación de córnea en ojo izquierdo. Rev Fac Med Univ Nac Colomb. 2004;52(3):223-9.
7. Chiaradia P. La córnea en apuros. Buenos Aires: Ediciones Científicas Argentinas; 2006.
8. American Academy of Ophthalmology. External disease and cornea. USA: American Academy of Ophthalmology; 2008. (Basic and Clinical Science Course)
9. De la Houssaye G, Bieche I, Roche O, Vieira V, Laurendeau I, Arbogast L, et al. Identification of the first intragenic deletion of the PITX2 gene causing an Axenfeld-Rieger syndrome: case report. BMC Med Genet. 2006;7:82.
10. Tümer Z, Bach-Hol D. Axenfeld Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. European Journal of Human Genetics. 2009 [citado 14 noviembre de 2010];17(12). Disponible en: http://www.nature.com/ejhg/journal/v17/n12/pdf/ejhg200993a.pdf
11. Tanwar M, Dada T, Dada R. Axenfeld-Rieger Syndrome associated with congenital glaucoma and cytochrome p4501b1 gene mutations. Case Report Med. 2010;2010 [citado 30 de diciembre 2010]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed?term=Axenfeld-RiegerSyndromeassociatedwithcongenitalglaucomaandcytochromep4501b1genemutations
12. Ayala E, Álvarez J, Delgado JL. Síndrome de Axenfeld-Rieger. Arch Soc Canar Oftal. 1999 [citado 30 de diciembre 2010];(10). Disponible en: http://www.oftalmo.com/sco/revista-10/sco17.htm
13. Oliveira M, Mitraud R, Yamane R. Axenfeld-Rieger anomaly and corneal endothelial dystrophy: a case series. Rev Bras Oftalmol. 2008;67(6):303-8.
14. Álvarez-Marín J, Delgado JL, Abreu P. Válvula de Ahmed en el glaucoma refractario. Primeros años de experiencia. Arch Soc Canar Oftal. 2005 [citado 30 de diciembre 2010];(16). Disponible en: http://www.oftalmo.com/sco/revista-16/16sco04a.htm
Recibido: 8 de marzo de 2012.
Aprobado: 8 de abril de 2012.
Dra. Yanileidy González Blanco. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave. 76 No. 3104 entre 31 y 41 Marianao. La Habana, Cuba. Correo electrónico: yani.glez@infomed.sld.cu
