










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión impresa ISSN 0864-2176
Rev Cubana Oftalmol vol.27 no.1 Ciudad de la Habana ene.-mar. 2014
PRESENTACIÓN DE CASO
Distrofia corneal de Schnyder
Schnyder crystalline dystrophy
MSc. Michel Guerra Almaguer,I MSc. Arelys Ariocha Cambas Andreu,II MSc. Carmen de Prada Sánchez,II MSc. Sol Inés Parapar Tena,II Dr. Lázaro Averoft,III Lic. Katia Lora DomínguezI
I Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
II Hospital "General Calixto García". La Habana, Cuba.
III Hospital "Freyre de Andrade". La Habana, Cuba.
RESUMEN
La principal entidad hereditaria con depósitos de lípidos en el estroma corneal es la distrofia cristalina central, conocida como distrofia de Schnyder, quien la describió en Suiza en 1927. Se caracteriza por depósitos blanco-amarillentos en el estroma corneal central y superficial. Se presenta un paciente de 28 años, del sexo masculino y piel negra, con antecedente de salud anterior. Acudió a consulta y refirió una disminución de la visión y cambio de coloración progresiva de ambos ojos, de años de evolución. En la exploración oftalmológica de ambos ojos se apreciaron lesiones blanquecinas anulares a nivel del estroma corneal, con ligera turbidez corneal central. Los estudios refractivos realizados constataron un astigmatismo hipermetrópico simple. El resto del examen oftalmológico fue negativo. Para el diagnóstico de certeza se empleó el microscopio confocal. Se concluye que el caso presenta una distrofia corneal estromal de tipo cristalina, de Schnyder.
Palabras clave: distrofia corneal de Schnyder, estroma corneal, microscopia confocal.
ABSTRACT
Central crystalline dystrophy known as Schnyder´s dystrophy, called like this because he was the first one to describe it in 1927 in Switzerland, is the main inherited entity with liquid deposits in the corneal stroma. This disease is characterized by white-yellow deposits into the central and superficial corneal stroma. A 28 years old black male patient, with previous history of health problems, went to the doctor´s office and mentioned visual reduction and progressive coloring changes in both eyes that had lasted some years. Ophthalmologic examination of both eyes showed annular white lesions at the corneal stroma, with light central corneal haze. Simple hypermetropic astigmatism was detected in the refractive exams. The rest of ophthalmologic exams was negative. For more secure diagnosis, confocal microscope was used. It was concluded that the patient had stromal corneal dystrophy, or Schnyder´s crystalline dystrophy.
Key words: Schnyder corneal dystrophy, corneal stroma, confocal microscope.
INTRODUCCIÓN
Las distrofias corneales continúan siendo enfermedades de complejo diagnóstico. En ocasiones es difícil distinguir las enfermedades de carácter auténticamente distrófico de otros trastornos congénitos causados por un defecto en el desarrollo embriológico.1 Se caracterizan por ser una alteración primaria y espontánea, bilateral y simétrica, de predominio central, de naturaleza no inflamatoria, de inicio precoz en la vida y progresión lenta, de origen hereditario, donde las células afectadas deben haber presentado inicialmente una función normal.2
La principal entidad hereditaria con depósitos de lípidos en el estroma corneal es la distrofia cristalina central, conocida como distrofia de Schnyder, quien la describió en Suiza en 1927. Se caracteriza por depósitos blanco-amarillentos en el estroma corneal central y superficial, aunque sin afectar el epitelio. Están formados por finos cristales policromáticos (colesterol y grasas neutras) entre las fibras de colágeno. Existen además cambios periféricos (arco lipoideo) y difusos de todo el estroma.3,4
La herencia de la distrofia cristalina central es autosómica dominante con alta penetración, y se ha encontrado en el gen UBIAD1 en 1P36, que afecta a uno y otro sexos con iguales probabilidades. La evidencia acumulada del espectro de mutación familiar de la distrofia corneal de Schnyder (SCD), la proteína mitocondrial UBIAD1, parece tener una función altamente conservada que, al menos en los humanos, está vinculada de manera noble con el metabolismo del colesterol.5-10
Aparece desde la 1ra. y la 2da. décadas de la vida y progresa lentamente. Puede presentar molestias importantes como fotofobia y pérdida de visión, sobre todo a partir de los 30 años. En edades más avanzadas puede asociarse a una forma pancorneal. La agudeza visual suele estar conservada si los depósitos respetan el eje visual. Se ha asociado con trastornos sistémicos del metabolismo lipídico (hiperlipidemia) a pesar de que esta asociación no es constante, y en un porcentaje variable de casos se encuentran valores normales de los lípidos plasmáticos. Además, puede aparecer genu valgum y condrodistrofia.3
Actualmente la fotoqueratectomía es muy efectiva para eliminar los depósitos superficiales y la queratoplastia tiene una tendencia a la recidiva mucho menor que en otras distrofias estromales.4
Por lo infrecuente que resulta esta entidad se decidió realizar la presentación del caso.
PRESENTACIÓN DEL CASO
Se presenta un paciente masculino, de 28 años de edad, coloración de la piel negra, quien acudió a consulta y refirió disminución de la visión y cambio de coloración progresiva de ambos ojos (AO), de años de evolución. Los antecedentes patológicos personales y oculares no fueron significativos; no se encontraron alteraciones al examen ocular en familiares de primera línea.
Al examen oftalmológico, los anexos y el fondo de ojo fueron normales. En el segmento anterior en la biomicroscopia en lámpara de hendidura se observaron lesiones blanquecinas anulares a nivel del estroma corneal, con ligero edema corneal central de ambos ojos (Fig. 1).
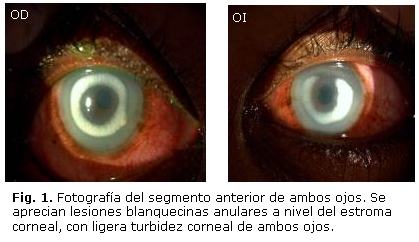
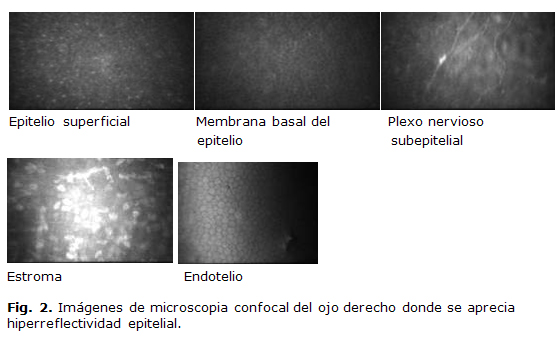
La tensión ocular era de 14 mmHg en el ojo derecho (OD) y 12 mmHg en el ojo Izquierdo (OI). La refracción y la agudeza visual mejor corregida era en el OD +1,25 x 100º (1,0) y en el OI +1,75 x 80º (1,0). La paquimetría central en el ojo derecho 611 micras y en el ojo izquierdo 616 micras. Como parte del estudio de la lesión corneal de ambos de ojos se procedió a realizar microscopia confocal (Fig. 2). Los estudios de lípidos en sangre no mostraron alteración; además, no se evidenciaron alteraciones osteomioarticulares.
DISCUSIÓN
La distrofia es considerada infrecuente, por lo que quizás muchos oftalmólogos nunca llegarán a examinar a un enfermo de (SCD). El diagnóstico y la comprensión de la SCD se hace aun más difícil por el número de artículos publicados que continúan desinformando sobre la enfermedad y que continúan enfatizando en la necesidad de demostrar la presencia de cristales estromales para llegar al diagnóstico, y solo en él la mitad de los pacientes afectos llegan a tener cristales corneales.11,12
El Instituto Nacional de Salud de Estados Unidos detalla la distrofia como una enfermedad poco frecuente con menos de 20 000 casos. Recientemente informó sobre el mayor estudio publicado de personas con SCD, en el que se describen 115 individuos afectados provenientes de 34 familias. El presente estudio, realizado entre 1987 y 2006, encontró esta enfermedad en blancos, occidentales y afroamericanos.11
Aunque la mayoría de los casos de SCD informados en la literatura son de origen caucásico, también se han reportado en pacientes orientales. No se han publicado artículos que reporten la ocurrencia de SCD en la población cubana. Se han manifestado casos en asiáticos, indios y sauditas.13,14
Se han reportado casos de genu valgum en personas con antecedentes de SCD. En el grupo de 115 enfermos estudiados, aproximadamente el 4 % de los casos presentaron genu valgum.11
Se han descrito numerosos patrones de distrofia cristalina de Schnyder,14 entre ellos: 1) una opacidad discoide central sin aspecto cristalino, 2) una opacidad cristalina discoide central con bordes festoneados 3) una opacidad cristalina discoide central con bordes mal definidos, 4) una opacidad cristalina anular con centro claro y 5) una opacidad anular con acúmulos cristalinos y centro claro.1,15
En cuanto al diagnóstico diferencial, es fundamental descartar degeneraciones lipoideas secundarias a queratitis intersticiales. En estas los depósitos lipídicos provienen de vasos estromales tras un proceso inflamatorio ocular crónico. En la distrofia de Schnyder no aparecen estos vasos estromales ni los surcos característicos de regresión de estos, además de no existir antecedentes de proceso inflamatorio crónico.
Podría confundirse también con la distrofia cristalina marginal de Bietti, en la que no solo existen depósitos de cristales de colesterol en la córnea, sino que también se hallan en la retina al ser ocasionado por un defecto sistémico en el metabolismo de los lípidos. Se deben descartar otras enfermedades sistémicas que pueden ocasionar depósito patológico de sustancias: disproteinemias como el mieloma múltiple y la macroglobulinemia de Waldenstrom (donde las inmunoglobulinas son el origen de los depósitos), la gota o hiperuricemia con cristales de uratos, las hiperbilirrubinemias de cualquier etiología con acúmulo de bilirrubina, y otras enfermedades de depósito de lípidos como la deficiencia LCAT (Lecitin Colesterol Acil-Transferasa), enfermedad de ojo de pez y enfermedad de Tangier. En todas estas enfermedades de depósito, otros tejidos, además de la córnea, se encuentran afectos por la acumulación patológica de las diversas sustancias.16,17
El tratamiento dependerá de los síntomas y de la agudeza visual. Cuando existe una opacidad capaz de reducir la visión de todo el espesor corneal estará indicada la queratoplastia, la cual tiene un pronóstico relativamente bueno, con tendencia a la recidiva mucho menor que en otras distrofias estromales.4
No se ha reportado nunca la periodicidad de intervenciones quirúrgicas de córneas en casos de SCD. Lo infrecuente de la distrofia hace que la mayoría de las publicaciones traten de casos reportados o de pequeñas series que describen agudeza visual en un número limitado de pacientes afectados.11
La mayoría de los artículos sobre la queratoplastia penetrante (PKP) en SCD refieren solo casos reportados, por lo que no existen recomendaciones en dicha literatura sobre cuándo indicar PKP en los pacientes con la enfermedad. Además, los casos de PKP en SCD a menudo carecen de datos importantes que permitan hacer pronósticos para cirugía. Por ejemplo, Weller y Rodger reportan que aplicaron PKP en «mujer cincuentona soltera que no podía realizar su trabajo», pero los autores no informaron sobre su visión antes del PKP.11 Hay reportes donde se refiere que la distrofia raramente requiere de trasplantes de córnea.
La queratectomía fototerapéutica (PTK) es una alternativa en el tratamiento de estos pacientes. Es capaz de mejorar las molestias oculares producidas por irregularidades epiteliales (irritación, sensación de cuerpo extraño, fotofobia), prevenir erosiones corneales, así como retrasar o evitar intervenciones corneales más agresivas, como la queratoplastia (lamelar o penetrante) en distrofias corneales superficiales con las posibles complicaciones que estas acarrean, como es la de un rechazo inmunológico.3,11,18
Se sabe que los mejores resultados visuales con PTK en este tipo de individuos se obtienen en aquellos pacientes en los que las lesiones son más superficiales. Es un procedimiento externo y ambulatorio que puede repetirse con facilidad en caso de recidiva. La principal desventaja se halla en la hipermetropización y la probabilidad de que ocurra Haze es muy excepcional.3,11,18
Varios estudios han demostrado una recurrencia baja de la enfermedad después del PTK; sin embargo, un número pequeño tiene recurrencia y necesita más de un tratamiento.3,11,18
A pesar de ser este tipo de distrofia infrecuente, el oftalmólogo debe tener presente las características clínicas para lograr un adecuado diagnóstico y tratamiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Martí Huguet T. Distrofias corneales. Barcelona: Cusí S.A.; 1996.
2. Eguía Martínez F, Ríos Torres M, Capote Cabrera A. Manual de diagnóstico y tratamiento en oftalmología. La Habana: Editorial Ciencias Médicas; 2009.
3. Orphanet. Portal de Información de enfermedades raras y medicamentos huérfanos. Distrofia corneal cristalina de Schnyder [Internet]. 2013 [citado 21 de octubre de 2013]. Disponible en: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=98967
4. Barraquer RI, Toledo MC, Torres E. Distrofia y degeneraciones corneales. Atlas y texto. Barcelona: Espaxs S.A.; 2004.
5. Weiss JS, Kruth HS, Kuvaniemi H, Tromp G, White PS, Winters RS, et al. Mutations in the UBIAD1 gene on chromosome short arm 1, region 36, cause Schnyder crystalline corneal dystrophy. Invest Ophthalmol Vis Sci. 2007;48(11):500-712.
6. Nickerson ML, Kostiha BN, Brandt W, Fredericks W, Xu KP, Yu FS, et al. UBIAD1 mutation alters a mitochondrial prenyltransferase to cause Schnyder corneal dystrophy. PLoS One [Internet]. 2010 [citado 12 enero de 2012];5(5):[aprox 42 p.]. Disponible en: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0010760
7. Jing Y, Liu C, Xu J, Wang L. A novel UBIAD1 mutation identified in a Chinese family with Schnyder crystalline corneal dystrophy. Mol Vis. 2009;15:1463-9.
8. Weiss JS, Kruth HS, Kuivaniemi H, Tromp G, Karkera J, Mahurkar S, et al. Genetic analysis of 14 families with Schnyder crystalline corneal dystrophy reveals clues to UBIAD1 protein function. Am J Med Genet A. 2008;146(3):271-83.
9. Yellore VS, Khan MA, Bourla N, Rayner SA, Chen MC, Sonmez B, et al. Identification of mutations in UBIAD1 following exclusion of coding mutations in the chromosome 1p36 locus for Schnyder crystalline corneal dystrophy. Mol Vis. 2007;13:1777-82.
10. Kobayashi A, Fujiki K, Murakami A, Sugiyama K. In vivo laser confocal microscopy findings and mutational analysis for Schnyder's crystalline corneal dystrophy. Ophthalmology. 2009;116(6):1029-37.
11. Weiss JS. Visual morbidity in thirty-four families with schnyder crystalline corneal dystrophy (an American Ophthalmological Society thesis). Trans Am Ophthalmol Soc. 2007;105:616-48.
12. Weiss JS, Moller HU, Lisch W, Kinoshita S, Aldave AJ, Belin MW, et al. La clasificación IC3D de las distrofias corneales. Córnea. 2008;27(Suppl. 2):43-83.
13. Du C, Li Y, Dai L, Gong L, Han C. A mutation in the UBIAD1 gene in a Han Chinese family with Schnyder corneal dystrophy. Mol Vis. 2011;17:2685-92.
14. Al-Ghadeer H, Mohamed JY, Khan AO. Schnyder Corneal Dystrophy in a Saudi Arabian Family with Heterozygous UBIAD1 Mutation. Middle East: Afr J Ophthalmol. 2011;18(1):614.
15. Krachmer JH, Mannis MJ, Holland EJ. Cornea. Philadelphia: Elsevier Mosby; 2005.
16. Lu LW. Distrofia estromal cristalina hereditaria de Schnyder. Rev Cibern Oftalmol. 2008:[aprox 5 p.]. Disponible en: http://www.oftalmologia.org/rco/index.php?option=com_content&view=article&id=234:distrofia-estromal-cristalina-hereditaria-de-schntder-eduardo-perez-salvador-md&catid=17:cornea&Itemid=36
17. Weiss JS, Khemichian AJ. Differential diagnosis of Schnyder corneal dystrophy. Dev Ophthalmol. 2011;48:67-96.
18. Cárdenas T, Capote A, Benítez MC, Castillo AC. Otras técnicas de cirugía corneal con el empleo de láser de excimer. II Taller de la Cátedra de Córnea y Cirugía Refractiva. I Jornada de la sección de cirugía refractiva, córnea y catarata de la Sociedad Cubana de Oftalmología; 2010.
Recibido: 20 de septiembre de 2012.
Aprobado: 14 de marzo de 2013.
Dr. Michel Guerra Almaguer. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave. 76 No. 3104 entre 31 y 41 Marianao, La Habana, Cuba. Correo electrónico: michguerra@infomed.sld.cu
