










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol vol.27 no.4 Ciudad de la Habana oct.-dic. 2014
INVESTIGACIÓN
Características clínicas de la distrofia corneal endotelial de Fuchs
Clinical characteristics of Fuchs' endothelial corneal dystrophy in families under study
Dra. Madelaine López González,I Dr. Urbano Rodríguez de la Paz,II Dra. Silvia María López Hernández,II Dra. Suzel Lapido Polanco,II Dr. Waldemar Baldoquin RodríguezIII
I Hospital Pediátrico Docente "Juan Manuel Márquez". La Habana, Cuba.
II Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
III Servicios Médicos de Cuba. La Habana, Cuba.
RESUMEN
Objetivos: caracterizar clínicamente la distrofia corneal endotelial de Fuchs en las familias estudiadas.
Métodos: se realizó un estudio observacional, descriptivo y transversal en familias con distrofia de Fuchs, en el Instituto Cubano de Oftalmología "Ramón Pando Ferrer", del año 2008 al 2011. Se operacionalizaron variables como: estadio clínico de la enfermedad, amplitud de cámara anterior y ángulo iridocorneal, presión intraocular, longitud axial, paquimetría, densidad celular endotelial, pleomorfismo y polimegatismo. Se resumieron las variables utilizando frecuencias absolutas y relativas porcentuales. Para la relación entre densidad celular y paquimetría se hizo un análisis de correlación bivariado no paramétrico.
Resultados: los pacientes enfermos se encontraban en su mayoría en el estadio I y II, con 50 y 60 % para los recién diagnosticados y los ya conocidos respectivamente. La amplitud de la cámara anterior fue normal en el 51,9 %; el ángulo iridocorneal estrecho en el 55,6 %, la presión intraocular normal en el 66,7 % y una hipermetropía axial en el 55,6 % de los casos con distrofia corneal. No se encontró asociación significativa entre paquimetría y densidad celular, pero sí pleomorfismo y polimegatismo en el 100 % de los enfermos y en más del 50 % de los descendientes directos.
Conclusiones: las variables analizadas demuestran ser importantes a la hora de caracterizar la enfermedad, además de señalar daños endoteliales en descendientes no enfermos, aspecto a tener en cuenta para su seguimiento.
Palabras clave: distrofia endotelial de Fuchs, distrofia corneal, endotelio corneal.
ABSTRACT
Objective: to describe the clinical characteristics of Fuchs' endothelial corneal dystrophy in the families under study.
Methods: observational, descriptive and cross-sectional study of families with Fuchs' corneal dystrophy diagnosed at "Ramón Pando Ferrer" Cuban Institute of Ophthalmology from 2008 to 2011. The study variables were clinical stage of the disease, depth of anterior chamber and iridocorneal angle, intraocular pressure, axial length, corneal thickness, endothelial cell density, pleomorphism and polymegatism. The variables were summarized using percentage absolute and relative frequencies. For assessing the ratio of the cell density and pachymetry, a nonparametric bivariate correlation analysis was made.
Results: the patients with Fuchs' endothelial corneal dystrophy were mostly in the clinical stages I and II, accounting for 50 % of the newly diagnosed patients and 60 % of the previously known ones. The amplitude of the anterior chamber was normal in 51,9 %, the iridocorneal angle was narrow in 55,6 %. Intraocular pressure was normal in 66,7 % and axial hypermetropia was found in 55,6 % of cases with corneal dystrophy. No significant association was found between the corneal thickness and cell density, but it was between pleomorphism and polyimegathism in 100 % of patients and in more than 50 % of the direct descendants.
Conclusions: the variables proved to be important in characterizing the disease as well as in pointing endothelial damages among non-sick descendants. This aspect should be considered on the follow-up period.
Key words: Fuchs's endotelial dystrophy, corneal dystrophy, corneal endothelium.
INTRODUCCIÓN
Tradicionalmente para el oftalmólogo las distrofias corneales son enfermedades complejas en su diagnóstico y diferenciación, consideradas como una alteración primaria y espontánea, casi siempre bilateral y simétrica, de predominio central, de inicio precoz en la vida y de progresión por lo general lenta.1
Ernst Fuchs utilizó por vez primera el término distrofia corneal (DC) a principios del pasado siglo, y a partir de este momento recibió denominaciones como distrofia corneal endotelial de Fuchs (DCEF) o simplemente distrofia de Fuchs. La córnea es la estructura más anterior del globo ocular, donde aparece esta afección, que a su vez está constituida por cinco capas: epitelio, capa de Bowman, estroma, membrana de Descemet (MD) y endotelio.1
La DCEF es un trastorno degenerativo específico y progresivo del endotelio corneal, donde la MD puede estar muy afectada con engrosamiento y excrecencias posteriores denominadas clínicamente gutas o verrugas, que se forman a partir de células endoteliales que secretan una matriz extracelular, la cual adquiere una compleja estructura laminar que se hace más gruesa con la edad, rica en colágeno tipo IV, tipo VIII, fibronectina y laminina.1,2-4
Al producirse discontinuidades en el endotelio fallan su función de barrera y la actividad de sus bombas iónicas, lo que crea insuficiencia en la resorción de fluidos y acumulación de agua en la córnea, con todo el cortejo sintomático consecuente.5,6
La DCEF afecta aproximadamente el 4 % de la población mayor de 40 años, y mucho más a mujeres que a hombres. Se ha encontrado asociado a condiciones como longitud axial (LA) disminuida, cámara anterior (CA) estrecha, glaucoma, queratocono, enfermedad cardiovascular y neuropatía óptica hereditaria de Leber; y aunque no abundan las investigaciones que impliquen familias enteras, la mayoría de ellos señalan una transmisión autosómica dominante (muchos casos clínicos parecen ser esporádicos). Los progresos en este campo se realizaron basados en estudios sobre el colágeno tipo VIII, y se detectaron mutaciones en el gen para su cadena a2 (COL8A2), en los brazos cortos del cromosoma 1.1,3-8-10
La fase inicial de esta enfermedad tiende a ser asintomática. Cuando aparece edema responde al tratamiento conservador al comienzo, hasta que produce cambios en la transparencia corneal con síntomas visuales significativos como deslumbramiento, déficit visual e incluso dolor en fases más avanzadas. Es la indicación frecuente de queratoplastia penetrante, o más recientemente endoqueratoplastia.1,11,12
La pirámide poblacional en Cuba está invertida; cada día nuestra población se vuelve más añosa, por lo que se hace más frecuente encontrar este tipo de afección corneal. En la medida en que conozcamos más datos clínicos sobre la enfermedad, estaremos en condiciones de diagnosticarla y tratarla precozmente, y este es el objetivo fundamental de nuestro estudio.
MÉTODOS
Se realizó un estudio observacional, descriptivo y transversal en 78 miembros de 15 familias trigeneracionales; cada familia parte de un paciente conocido con DCEF seguido en consulta de córnea del Instituto Cubano de Oftalmología "Ramón Pando Ferrer", del año 2008 al 2011, de las que se desglosan 63 descendientes directos.
Se tomaron en cuenta como criterios de inclusión: que tuvieran tres generaciones vivas partiendo del paciente de forma ascendente o descendente; tuvieran más de 10 años de edad; sin antecedentes oftalmológicos previos de traumas, inflamaciones oculares, usuarios de lentes de contacto o de intervención quirúrgica del segmento anterior y que aceptaran participar en el estudio.
Se confeccionaron planillas de recolección y se tomaron los resultados de la base de datos del equipo de microscopio confocal (Confoscan 4 NIDEK Technology), donde se le realizó la microscopIa confocal (MC) a cada ojo de pacientes y familiares.
Se operacionalizaron las siguientes variables:
-Estadio clínico de la DCEF: para la clasificación de cada paciente dentro de estas categorías se tomó el estadio correspondiente al ojo más afectado (usada a nivel internacional):1
Para el análisis del resto de las variables también se tomaron las medidas del ojo más afectado, excepto en los casos de estadio IV. En los casos de afectación corneal simétrica se tomó un ojo al azar en el que fue analizado lo siguiente:
- Amplitud de la cámara anterior: obtenida mediante biometría, llevados a escala cualitativa.
-Paquimetría: medida del grosor corneal expresada en micras (µ).1,14,15 Se obtuvo mediante MC.
-Pleomorfismo: células endoteliales que mantienen su forma hexagonal, obtenida mediante MC, llevada a escala cualitativa.
Los datos recogidos fueron pasados a una base de datos (Excel) para su procesamiento estadístico. Se resumieron las variables cualitativas utilizando frecuencias absolutas y relativas porcentuales. Para determinar la relación entre densidad celular y paquimetría se hizo un diagrama de dispersión y análisis de correlación bivariadas no paramétrico con la ayuda del coeficiente de correlación de Spearman, con un nivel de confiabilidad de 0,05 según Softwear estadístico SPSS 15.0. Los participantes recibieron información detallada sobre el estudio y firmaron el consentimiento informado, cumpliendo el código de ética para las investigaciones biomédicas.
RESULTADOS
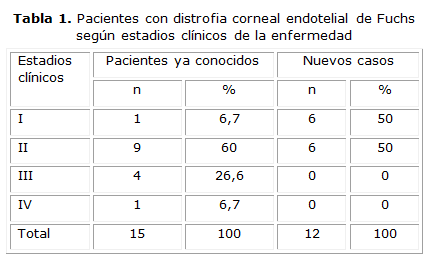
Se determinó la presencia de la enfermedad en 12 descendientes directos (considerados como nuevos casos), donde los estadios I y II de la enfermedad se encontraron con igual frecuencia, para un 50 % de presentación. A su vez, el estadio II fue el más observado en el grupo con diagnóstico previo, con 9 pacientes para un 60 % (tabla 1).
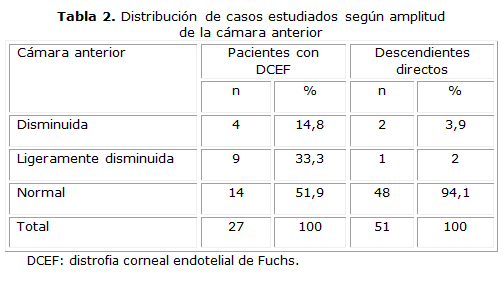
Los nuevos casos descendientes directos de enfermos ya conocidos con DCEF se consideraron como pacientes con DCEF a partir de ese momento, por lo que aumentó a 27 el total de pacientes con la enfermedad corneal. En la tabla 2 se observa que 13 de los pacientes con DCEF presentaron una CA entre ligeramente disminuida y disminuida, que representó el 48,1 % de los casos dentro de su grupo. La mayoría de los descendientes directos presentaron CA normal.
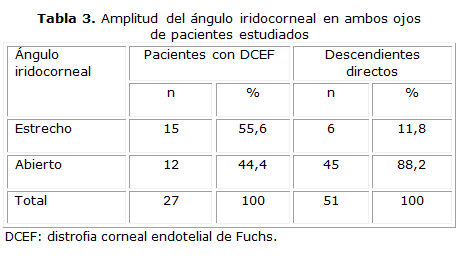
Más de la mitad de los casos con DCEF presentaron ángulo estrecho (55,6 %); sin embargo, el 88,2 % de los descendientes directos tenían ángulo abierto (tabla 3).
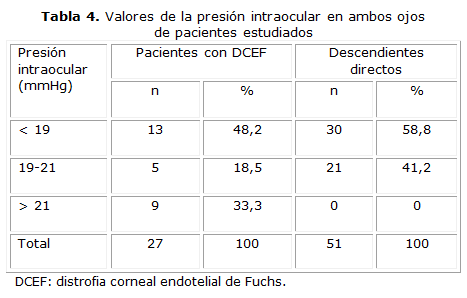
Tanto en el grupo con enfermedad corneal diagnosticada como en sus familiares predominaron las cifras de PIO inferiores a los 19 mmHg, con un 48,2 % de los casos para el primer grupo y 58,8 % en el segundo. Cifras superiores a los 21 mmHg solo fueron constatadas en pacientes con DCEF (tabla 4).
Como se muestra en la tabla 5, el grupo con DCEF presentó hipermetropía axil en 15 pacientes para un 55,6 %. En los descendientes directos predominó la LA dentro de límites normales con 46 pacientes para un 90,2 %.

Al analizar los valores de paquimetría y densidad celular en pacientes con DCEF, se determinó que estos no mostraron un patrón de asociación de manera significativa. Este resultado se corroboró con la prueba de correlación no paramétrica (Rho de Spearman= 0,011, p= -0,348 significativo) a un nivel de confianza de 0,05 (Fig.).

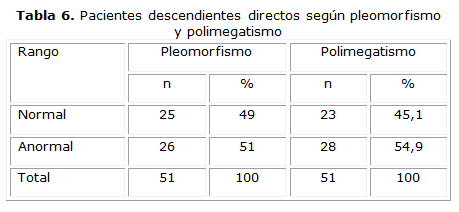
Todos los pacientes con DCEF presentaron alteraciones en el pleomorfismo y polimegatismo celular, lo que representa el 100 % de los casos. En la tabla 6 solo está representado el comportamiento de estos parámetros en pacientes descendientes directos, donde el 51,0 % presentó alteraciones en la forma de la célula endotelial y el 54,9 % en el tamaño.
DISCUSIÓN
El diagnóstico de DCEF rara vez se encuentra en duda cuando se hace un correcto y detallado examen clínico, pero la estratificación por estadios es más subjetiva, por lo que se hace necesario tener otros elementos en cuenta para determinar el grado de severidad de la enfermedad y así poder intervenir prontamente. No es extraño además encontrar nuevos casos en estas familias si se tiene en cuenta el patrón de herencia autosómico dominante que prima en esta enfermedad.16
En los pacientes conocidos con DCEF los estadios I y II fueron los más frecuentes, lo que es de esperar si se tiene en cuenta que en etapas más avanzadas de la enfermedad suele ser el tratamiento quirúrgico la solución, y por lo tanto no se ajustan a los criterios de inclusión del presente estudio; sin embargo, en los descendientes directos con reciente diagnóstico de la enfermedad, las etapas antes mencionadas también fueron las más frecuentes, lo que guarda relación con las características clínicas de estas dos etapas, donde es posible que los pacientes se encuentren asintomáticos o tengan pocos síntomas visuales, atribuibles por ellos mismos en ocasiones a defectos refractivos o a fatiga visual, por lo que no acuden al especialista. Al ser una enfermedad de larga evolución, los pacientes asisten cuando presentan más síntomas y se corresponde entonces con etapas más tardías de la distrofia.1,2,17
Los pacientes con DCEF presentaron CA disminuida en un porcentaje muy cercano a la media, así como ángulo estrecho y LA disminuida en más de la mitad de sus casos; sin embargo, la PIO se mantuvo por debajo de los límites catalogados a nivel mundial como normales tanto en los enfermos como en los descendientes no afectados.7,8
Algunos autores describen la asociación de esta distrofia a CA y LA disminuida, así como a glaucoma, pero no se ha encontrado la verdadera justificación a estos eventos. Se plantea la presencia del edema corneal resultante del proceso evolutivo de descompensación de la distrofia como factor desencadenante que al incrementar el grosor de esta estructura pudiera interferir en la presencia de CA estrecha o ángulo estrecho, y provocar entonces la elevación de la PIO y el glaucoma subsecuente.7,8
El endotelio corneal está compuesto por una única capa de células hexagonales con uniones fuertes entre sí que cumplen la función de controlar el paso de fluido dentro y fuera de la córnea con el objetivo fundamental de mantener la transparencia corneal además de su nutrición y para esto es necesario que se mantenga un número adecuado de estas que puedan al menos suplir la función de aquellas que con el paso de los años, por mecanismos fisiológicos o no, hayan desaparecido.2,6,15,18
La guía de buenas prácticas de la Academia Americana de Oftalmología sugiere riesgo significativo de edema corneal, sobre todo para la cirugía de catarata cuando el conteo celular es menor de 1 000 células/mm2; sin embargo, otros autores han descrito córneas claras con conteos menores a las 500 células/mm2.1,2,17
La densidad celular y la paquimetría fueron analizadas en nuestra investigación buscando la posible relación entre estas variables. Resultó no significativa la relación entre estas, aunque queda claro que fisiológicamente a medida que disminuye la densidad celular puediera observarse un discreto aumento del grosor corneal.1,2,6,14,15,19
Según lo antes expuesto y teniendo en cuenta los mecanismos fisiológicos de la córnea para mantener su deturgencia, las células sufren una serie de cambios en su forma y tamaño al suplir el espacio de aquellas que desaparecen, por lo que es lógica la presencia de pleomorfismo y polimegatismo anormal en toda la serie estudiada de pacientes con DCEF; sin embargo, en los descendientes directos de estos que están exentos de enfermedad se observó la anormalidad de ambas variables en más del 50 % de los casos.
Burns y otros estudiaron la relación entre pleomorfismo y polimegatismo con la hidratación corneal como medida de la función celular endotelial, y encontraron relación con respecto a la variación en el tamaño, no así con la variación de la forma donde no fue importante.18
El estudio clínico de pacientes con sintomatología aislada de DCEF quizás no es tan importante desde el punto de vista preventivo como cuando se estudia la familia de un paciente que ya tiene el diagnóstico, ya que en los primeros acudirían cuando la enfermedad corneal ya está instalada, mientras que los segundos se pesquisarían sin síntomas, y darían más tiempo al seguimiento y tratamiento precoz de estos pacientes con el fin de evitar mayores daños que desencadenan pérdida visual a largo plazo.
Se concluye que las variables analizadas demuestran ser importantes a la hora de caracterizar la enfermedad, y se señala la presencia de daños endoteliales en descendientes no enfermos, aspecto a tener en cuenta para su seguimiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Barraquer RI, De Toledo MC, Torres E. Distrofias y degeneraciones corneales. Atlas y Texto. Barcelona: Expas; 2004. p. 209-21.
2. Rodríguez MM, Krachmer JH, Hackett J. Fuchs' corneal dystrophy. A clinicopathologic study of the variation in corneal edema. Ophthalmology. 1986;93:789-96.
3. Laing R. High magnification techniques. En: Krachmer JH. Cornea. Fundamentals of cornea and external disease. St Louis: Mosby. 1997:313-34.
4. Murphy C, Alvarado J, Juster R. Prenatal and postnatal growth of the human Descemet's membrane: prenatal and postnatal growth of human Descemet's membrane. Invest Ophthalmol Vis Sci. 2004;25:1402-15.
5. Hecker AL, McLaren JW, Bachman LA, Sanjay V, Patel BA. Anterior Keratocyte Depletion in Fuchs's Endothelial Dystrophy. Arch Ophthalmol. 2011;129(5):555-61
6. Mok JW, Kim HS, Joo CK. Q455V mutation in COL8A2 is associated with Fuchs's corneal dystrophy in Korean patients. Eye. 2009;23:895-903.
7. Louttit MD, Kopplin LJ, Igo RP Jr, et al. FECD Genetics Multi-Center Study Group. A multicenter study to map genes for Fuchs's endothelial corneal dystrophy: baseline characteristics and heritability. Cornea 2012;31:26-35.
8. Herndon LW. New visual function technologies in glaucoma. En: Shields MB (Ed). Defining glaucoma. Diagnosis and management of glaucoma. New York: Columbia University College of Physceant and surgeon; 2004:123-34.
9. Biswas S, Munier FL, Yardley J. Missense mutation in COL8A2, the gene encoding the a2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2006;10:2415-23.
10. Aldave AJ, Rayner Sa, Salem AK. No pathogenic mutations identified in the COL8A2 genes in familial with Fuchs's corneal dystrophy. Invest Ophthalmol Vis Sci. 2006;47(9):3787-90.
11. Azard DT, Jain S, Sambursky R, Strauss L. Microkeratome-assisted posterior keratoplasty. J Cataract Refract Surg. 2004;27:1903-4.
12. Ishino Y, Sano Y, Nakamura T, Endo K, Tsuzuki M, Tanifuji N, et al. Amniotic membrane as carrier for cultured corneal endothelium in vitro. Invest Ophthalmol Vis Sci. 2004;18:854.
13. Robert L, Roderick R, Huntington F. Patterns of single-gene inheritance. En: Thompson & Thompson. Genetics in Medicine. Philadelphia: Saunders. 2005:345-67.
14. Giasson CJ, Graham A, Blouin JF. Morphometry of cells and guttae in subjects with normal or guttae endothelium with a contour detection algorithm. Eye Contact Lens. 2005;31(4):158-65.
15. Szentmary N. Epithelial cell, keratocyte and endotelial cell apoptosis in Fuchs's dystrophy. Eur J Ophthalmol. 2005;15(1):17-22.
16. Gottsch JD, Eghrari OA. Fuchs's corneal dystrophy. Expert Rev Ophthalmol. 2010;5(2):147-59.
17. Okumura N, Koizumi N, Ueno M, et al. Enhancement of corneal endothelium wound healing by Rho-associated kinase (ROCK) inhibitor eye drops. Br J Ophthalmol. 2011;95:1006-9.
18. Burns RR, Bourne WM, Brubaker RF. Endothelial function in patients with cornea guttata. Invest Ophthalmol Vis Sci. 2001;20:77-85.
19. Graves E. El edema corneal. Una complicación que puede ser evitada. En: Centurión V. El Libro del cristalino de las Américas. Brasil: Livraria Santos; 2007. p. 613-7.
Recibido: 28 de julio de 2014.
Aprobado: 15 de octubre de 2014.
Dra. Madelaine López González. Hospital Pediátrico Docente "Juan Manuel Márquez". Marianao, La Habana, Cuba. Correo electrónico: madelopezg@infomed.sld.cu
 Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
